

Alzheimer’s Inflammation: Treating a Fire We Still Can’t Measure
The MINDFuL trial: A Case Study in the Missing Inflammation Biomarker

Alzheimer’s disease is usually described through amyloid and tau. But a third player has moved into the center of the conversation: brain inflammation.
If the Alzheimer’s brain is inflamed, should we reduce inflammation? First, we need to answer a more basic question:
How do we know which patient actually has harmful brain inflammation?
That is the main problem this post is about.
The field is moving toward immune-based treatments before we have reliable tools to identify the right immune state in the right patient. The recent XPro1595 trial is a useful case study. It is not just a trial of one drug. It is a test of whether Alzheimer’s research is ready for precision neuroinflammation trials.
The Main Problem: We Lack a Good Brain Inflammation Marker
In Alzheimer’s disease, we now have increasingly useful biomarkers for amyloid and tau. They are not perfect, but they tell us something fairly specific about Alzheimer’s pathology.
Inflammation is harder.
We do have good markers of general inflammation. CRP, for example, is a useful blood marker. If CRP is high, it tells us that the body may be in an inflammatory state.
But CRP does not tell us where the inflammation is coming from.
A high CRP could reflect arthritis, infection, obesity, heart disease, gum disease, poor sleep, or many other conditions outside the brain. It may matter for brain health, but it does not prove that the Alzheimer’s brain itself is inflamed.
The same problem applies to ESR and HbA1C. They may capture body-wide inflammation, metabolic stress, or vascular risk. But they do not measure brain inflammation directly.
APOE4 is more closely tied to Alzheimer’s biology and may shape immune vulnerability in the brain. But APOE4 is a genetic risk marker, not a real-time inflammation test.
This creates a major problem for trials.
If we cannot measure brain inflammation well, then we may not know who should receive an immune-targeted treatment. We also may not know whether the treatment worked biologically, even if the clinical results move in the right direction.
In drug development, that matters.
A proper Phase 2 trial should do more than look for a clinical signal. It should help define the population, the dose, the target, and the biological readout. It should show target engagement: evidence that the drug hit the pathway it was designed to hit.
There Is Inflammation in the Alzheimer’s Brain. But Is It Good or Bad?
There is strong evidence that the brain’s immune system is active in Alzheimer’s disease.
In brain tissue, immune cells cluster around amyloid plaques. Genetic studies point toward immune pathways, including TREM2, CD33, INPP5D, PLCG2, and APOE.
APOE4, the strongest common genetic risk factor for late-onset Alzheimer’s disease, is especially important here. It is not only about cholesterol. In the brain, APOE4 may affect lipid handling, amyloid clearance, blood-brain barrier function, and immune tone.
Single-cell studies also show that microglia can shift into disease-associated microglia, or DAM. DAM are often found near amyloid plaques. Early on, this may be protective: surrounding plaques, clearing debris, and containing injury.
But a protective response can become costly if it is pushed for too long.
In an APOE4 brain, where amyloid handling and immune signaling may already be stressed, DAM activation may be harder to sustain. Over time, a cleanup response may become less efficient, more inflammatory, or exhausted.
So the question is not whether inflammation exists in Alzheimer’s disease.
It does.
The better question is whether that inflammation is protective, harmful, or failing.
The Stage-Specific Model
One way to think about Alzheimer’s inflammation is by stage.
Early in the disease, inflammation may be mostly reactive and compensatory. The brain sees amyloid, damaged synapses, or cellular stress, and immune cells respond. In this setting, more immune activity may sometimes help.
This was the logic behind TREM2 agonists: enhance microglial cleanup and improve the brain’s response to amyloid. But recent TREM2 agonist trials have been disappointing, reminding us that boosting immune activity is not automatically helpful.
Later in the disease, the immune response may change. It may become chronic, senescent, or exhausted. The cells are still activated, but they may no longer be solving the problem. Instead of clearing damage, they may keep sending alarm signals and contribute to synapse loss.
This creates a different treatment goal.
Early disease may require supporting protective immune responses.
Later or inflammation-heavy disease may require inhibiting or re-aligning harmful immune signals.
This is why “anti-inflammatory treatment” is too blunt a phrase. The better goal may be stage-specific immune rebalancing.
The MINDFuL trial and the rationale for Soluble TNF
TNF, or tumor necrosis factor, is an immune signaling molecule. It is not automatically bad. The body and brain use TNF signaling for immune defense, repair, and communication between cells.
But TNF comes in different forms.
Soluble TNF can move through tissue and amplify inflammatory signaling.
Transmembrane TNF stays attached to cells and may help support normal immune balance, repair, and protective glial function.
This distinction is the rationale for XPro1595. The drug was designed to block soluble TNF while preserving transmembrane TNF. Mechanistically, XP is a dominant-negative TNF biologic. It binds to soluble TNF and forms inactive complexes, preventing soluble TNF from properly activating its receptors.
That is a reasonable biological idea. But it also raises a hard question:
Do we know which Alzheimer’s patients actually have excess soluble TNF signaling in the brain?
Right now, that answer is not clear.
XPro1595 is not really a DAM drug. It is not simply an immune exhaustion drug either. It is a soluble-TNF pathway drug. It assumes that, in some Alzheimer’s patients, inflammatory signaling has become maladaptive and that blocking soluble TNF may restore a healthier immune balance.
That assumption needs biomarkers.
What Justified Testing XPro1595?
The rationale for testing XPro1595 had three parts.
First, the biology of TNF suggested that selective soluble TNF blockade might be safer and more precise than broad TNF suppression.
Second, preclinical studies suggested that soluble TNF blockade could reduce glial activation, improve debris clearance, and restore synaptic plasticity.
Third, an earlier Phase 1b Alzheimer’s study reportedly showed dose-dependent reductions in cerebrospinal fluid biomarkers of neuroinflammation and neurodegeneration.
That last point is important because cerebrospinal fluid is closer to the brain than blood. But there is a caveat: those Phase 1b data were described as a manuscript in preparation. That means readers cannot fully judge the sample size, the biomarkers, the exposure-response relationship, or whether soluble TNF itself was measured.
So the rationale for MINDFuL was reasonable.
But it was not airtight.
What MINDFuL Tested
The MINDFuL trial was a Phase 2, randomized, double-blind, placebo-controlled study in early Alzheimer’s disease.
Participants had mild cognitive impairment or mild Alzheimer’s dementia. Treatment lasted 24 weeks. XPro1595 was given once weekly by subcutaneous injection.
The primary endpoint was EMACC, a cognitive composite designed to detect change in early Alzheimer’s disease.
That endpoint makes sense for Phase 2 signal detection. But it also has limits. EMACC is not the same as showing clear benefit on a more familiar clinical endpoint such as CDR-SB, which connects cognition and daily function.
The overall trial did not meet the primary endpoint-Figure below. Nillsh..
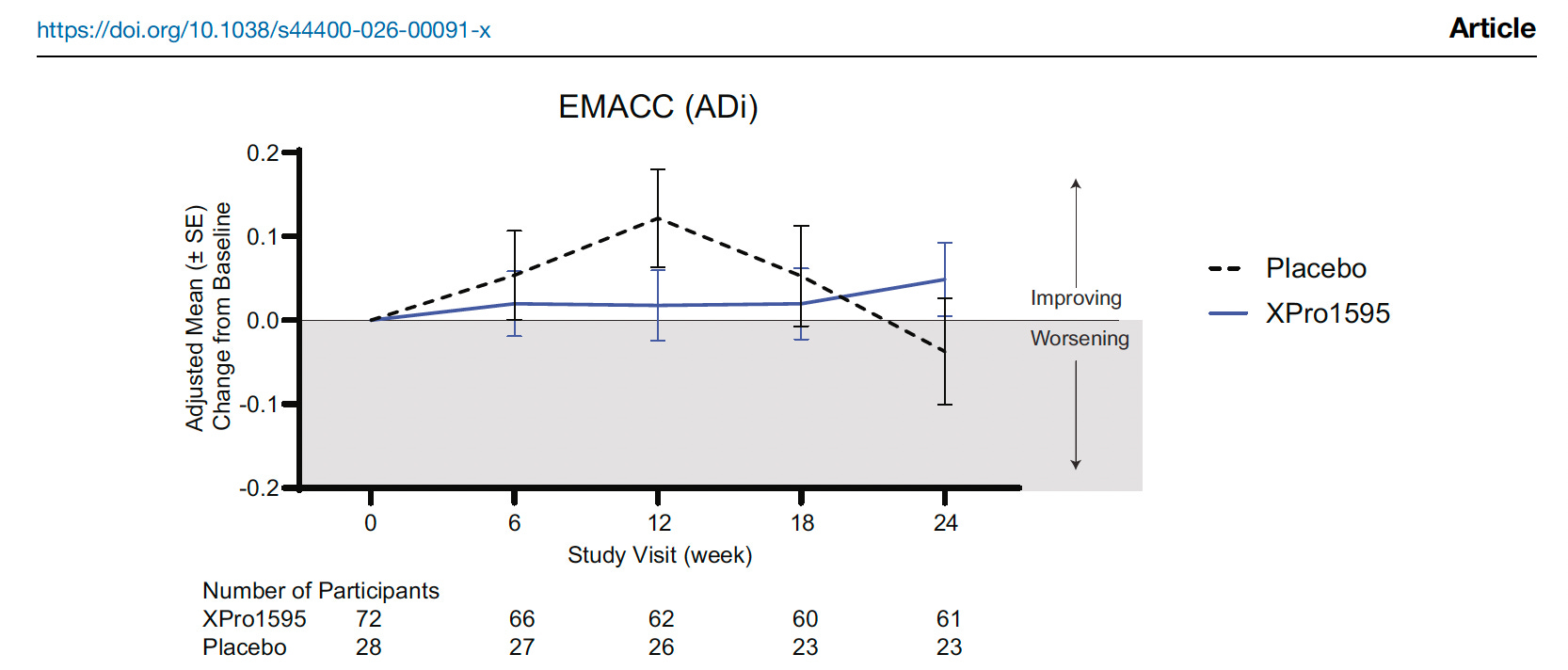
The study then focused on a subgroup called ADi, meaning Alzheimer’s disease with inflammation. These participants were amyloid-positive and had at least two inflammation-related markers.
The Core Critique
The core critique is not that XPro1595 had no rationale. It did.
The core critique is that the trial moved into Phase 2 without a sufficiently clear way to define the inflammatory population or prove target engagement in the brain.
The ADi subgroup was defined using CRP, ESR, HbA1C, and APOE4.
These markers may enrich for risk. They may identify people with systemic inflammation, metabolic stress, APOE4 biology, or faster expected decline.
But they do not prove harmful brain inflammation.
They also do not prove excess soluble TNF signaling in the central nervous system.
That distinction matters.
If the drug targets soluble TNF, then the ideal Phase 2 trial would show several linked steps:
The selected patients had evidence of relevant brain inflammation.
The drug reached the central nervous system.
The drug engaged soluble TNF signaling.
That engagement changed downstream biomarkers.
Those biomarker changes tracked with clinical benefit.
MINDFuL showed possible downstream biological activity, especially through p-tau217 and GFAP signals in the ADi subgroup. But these changes are not convincing for target engagement, even in the dose compliant ADi, an effect size of 0.26-0.22 is modest at best (figure below).
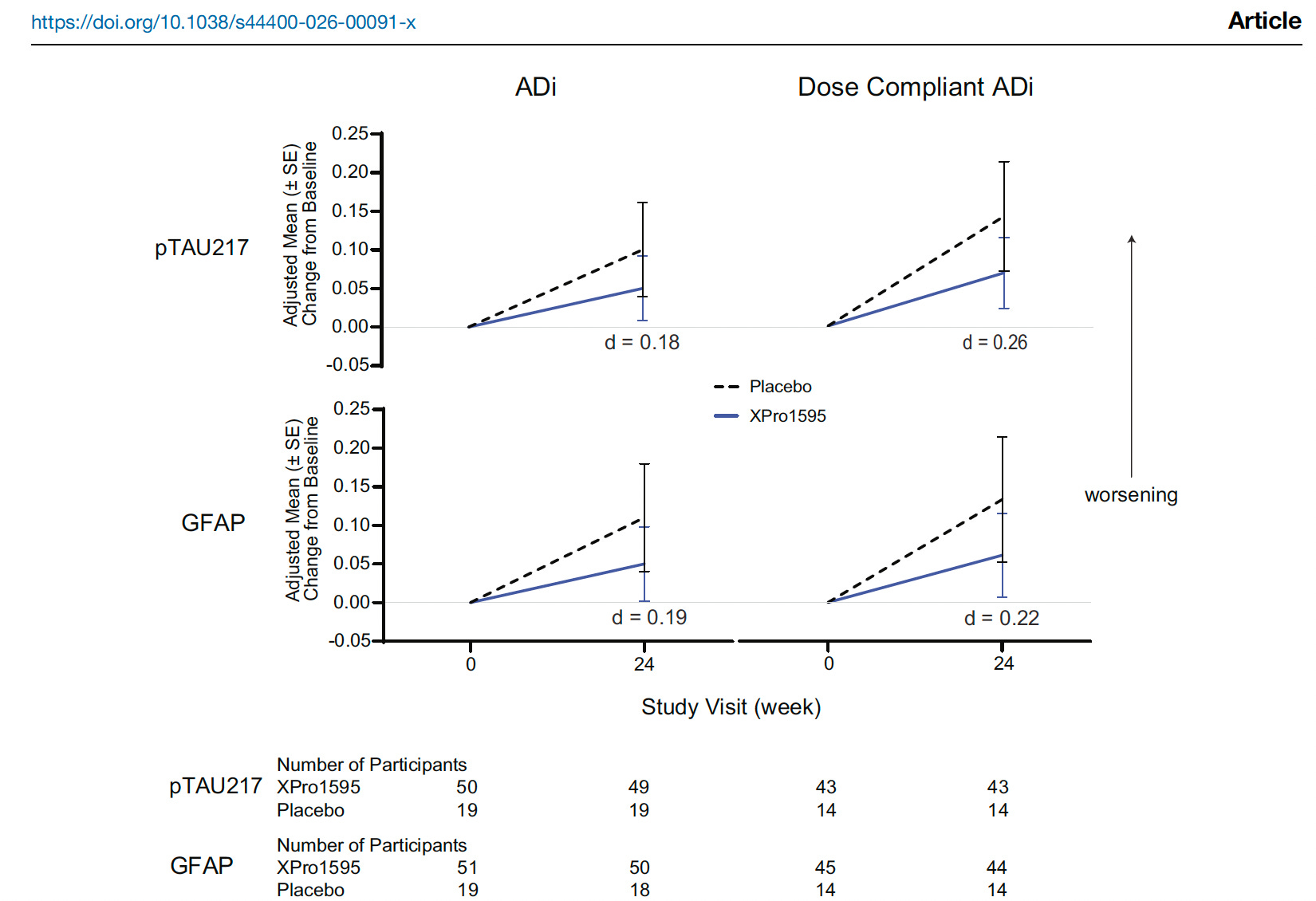
What do we do with these results?
Better Markers of Brain Inflammation
If this field is going to move forward, we need better biomarkers.
GFAP is one useful blood marker, but as seen above, not exactly responding to inhibition of soluble TNF. It reflects astrocyte activation and is closer to brain biology than CRP or ESR. But GFAP does not tell us which inflammatory pathway is active. It does not prove soluble TNF signaling. And worse, participants were not selected or stratified by baseline GFAP.
PET imaging may help. Some tracers, such as TSPO PET, aim to measure microglial activation. These are more brain-specific than CRP, but they still have limitations. TSPO is not exclusive to one cell type, tracer performance varies, and genetic differences can affect binding.
Experimental PET approaches may also help measure brain lipid biology.
Arachidonic acid, or AA, is involved in inflammatory lipid signaling. DHA, an omega-3 fatty acid, is important for brain membranes and inflammation resolution. AA and DHA PET are not routine clinical tools, but they point to an important idea: unresolved inflammation may involve lipid signaling, membrane turnover, and failed resolution, not just cytokines.
The field needs biomarkers that can distinguish:
body-wide inflammation
astrocyte activation
microglial activation
lipid inflammatory signaling
soluble TNF pathway activity
protective versus harmful immune states
Without this, we are partly guessing.
The Uncertainty
This is where the field needs humility.
Inflammation in Alzheimer’s may be protective early and harmful later.
Peripheral inflammation is not the same as brain inflammation.
APOE4 may increase inflammatory vulnerability, but it is not a real-time inflammation measure.
GFAP, PET imaging, and lipid PET approaches may improve the picture, but none is yet a perfect clinical tool.
Most importantly, we do not yet have a routine biomarker showing that a patient has excess soluble TNF signaling in the brain.
So XPro1595 remains scientifically interesting. But MINDFuL does not prove that we know how to identify the right patients or measure treatment efficacy with enough precision.
The Bigger Lesson
The future of Alzheimer’s inflammation treatment may not depend only on better drugs.
It may depend on better measurement.
The right patient.
The right disease stage.
The right immune pathway.
The right biomarker.
The right timing.
XPro1595 may or may not become an approved Alzheimer’s treatment. But it already teaches an important lesson:
A precision immune therapy requires precision immune biomarkers.
Without them, we risk running trials in vaguely defined populations and interpreting subgroup signals without knowing whether the drug reached the brain, hit the target, or treated the biology we intended to treat.
Take-Home Messages
The main barrier to targeting inflammation in Alzheimer’s disease is not only treatment. It is measurement.
We have useful markers of general inflammation, such as CRP, but we lack reliable markers of brain inflammation.
A blood marker can tell us that the body is inflamed without proving that the brain is inflamed.
Inflammation in Alzheimer’s may be protective early, harmful later, or ineffective when the immune response becomes exhausted.
APOE4 may shape brain immune vulnerability, but it is not a real-time inflammation test.
A precision immune therapy requires precision immune biomarkers.
A strong Phase 2 trial should show more than a clinical signal. It should show brain penetration, target engagement, biomarker change, and clinical benefit.
Without better biomarkers, Alzheimer’s neuroinflammation trials risk enrolling vaguely defined populations and producing results that are hard to interpret.
We will likely need multiple blood-based biomarkers to detect protective or harmful inflammation markers, and senescent markers may offer clues into the latter.
Key References
Jaeger J, Staats KA, Barnum S, et al. XPro1595 in early Alzheimer’s disease with inflammation: results from the phase 2 MINDFuL trial. npj Dementia. 2026.
Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurology. 2015.
Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer’s disease. Journal of Cell Biology. 2018.
Keren-Shaul H, Spinrad A, Weiner A, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017.
Bellenguez C, Küçükali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nature Genetics. 2022.
Vitek MP, Brown CM, Colton CA. APOE genotype-specific differences in the innate immune response. Neurobiology of Aging. 2009.
Ising C, Venegas C, Zhang S, et al. NLRP3 inflammasome activation drives tau pathology. Nature. 2019.
Reviews on TSPO PET, GFAP, and brain lipid metabolism imaging may be useful additions for readers who want a deeper dive into neuroinflammation biomarkers.
Thank you for your article. An interesting read. We are so far away from practicing true precision medicine.
Nice post Dr. Yassine! I was wondering if you think other imaging techniques in addition to PET, such as fMRI and DTI-ALPS, could also have applications in helping us with measurements in this context?