

Meet the Brain's Immune System — and Why It's the Next Big Bet Against Alzheimer's
A drug designed to supercharge the brain's immune cells just failed its biggest clinical test; but all is not lost.

The Brain Has a Cleanup Crew — and It Matters More Than We Thought
Your brain is not a passive organ. Tucked between neurons, constantly on patrol, is a population of cells called microglia — the brain’s resident immune cells. Think of them as a hybrid between a security guard and a janitor. When something goes wrong — a dead cell, a misfolded protein, a threatening invader — microglia are the first responders. They sense the problem, move toward it, surround it, and break it down.
In Alzheimer’s disease, one of the earliest events is the accumulation of a sticky protein called amyloid, which clumps into plaques outside neurons. Microglia are supposed to recognize those plaques and clear them. For decades, most Alzheimer’s research focused almost exclusively on amyloid itself, as if the cleanup crew didn’t exist. That changed in 2013.
A Signal Hidden in the Genome
That year, two independent research teams published back-to-back papers in the New England Journal of Medicine, each arriving at the same unexpected finding. They had been scanning the entire genetic code of tens of thousands of people — comparing those with Alzheimer’s to those without — searching for places where the DNA spelling differed. This kind of large-scale comparison, called a genome-wide association study or GWAS, is like comparing two manuscripts letter by letter across three billion characters, looking for the discrepancies that explain why one reader ends up with dementia and another doesn’t.
Both teams kept landing on the same gene: TREM2. A specific variant — called R47H — increased the risk of developing Alzheimer’s by two to four times. TREM2 encodes a receptor that sits on the surface of microglia, acting as the sensor that tells them when to swing into action. When TREM2 detects amyloid, cellular debris, or the molecular fingerprints of a dying cell, it instructs the microglia to cluster around the threat and clear it. TREM2, in short, is the alarm system of the brain’s cleanup crew.
A Tempting Hypothesis — and Its Early Warning Signs
The R47H variant blunts that alarm. It is a loss-of-function mutation: the sensor becomes less sensitive. People carrying it have microglia that respond sluggishly to amyloid plaques. Their brains show less microglial clustering around plaques, more diffuse and toxic forms of amyloid, and worse damage to nerve cell branches. Less TREM2 activity. More disease.
The therapeutic idea writes itself: if a broken alarm means the janitors don’t show up, what if you make the alarm louder? Could boosting TREM2 activity help the brain clear amyloid and slow the disease?
The first warning signs were easy to miss. One study found that chronically activating TREM2 could actually worsen the spread of tau — the second major toxic protein in Alzheimer’s, which propagates between neurons and drives irreversible neurodegeneration. Another comprehensive analysis tested TREM2-activating antibodies across multiple mouse models and found neutral or even detrimental results. But the genetic rationale was compelling enough that a clinical-grade drug was built, and a major trial was launched.
Building AL002: The Drug and Its Preclinical Story
What the drug does
AL002, developed by Alector, is a monoclonal antibody — an engineered protein designed to bind to TREM2 and activate it. When it binds, the receptor gets pulled inside the cell and degraded, triggering a burst of downstream signaling. A fragment of TREM2 that normally floats in spinal fluid — called soluble TREM2, or sTREM2 — drops sharply after treatment. Counterintuitively, that drop means the drug is working: the receptor was activated, did its job, and was cleared.
Why the animal studies were misleading
In mice engineered to carry both human TREM2 and five familial Alzheimer’s mutations — the 5xFAD model — a related antibody caused microglia to multiply around plaques, reduced the most toxic forms of amyloid, and decreased nerve cell damage. The mice looked better.
But several important gaps were quietly baked into this evidence. The molecule tested in mice was not AL002 itself — it differed in the region of the antibody that interacts with the broader immune system. The actual human drug was never tested in animals. The 5xFAD mice also accumulate amyloid at extraordinary speed driven by five stacked mutations borrowed from rare inherited forms of disease — an engineered catastrophe that bears little resemblance to the decades-long, genetically complex disease unfolding in a 70-year-old patient. And the mice were treated early, before significant pathology had accumulated, while patients in the trial already had confirmed amyloid and measurable cognitive impairment. Testing a fire suppression system in a controlled drill, then expecting it to work the same way in a building already fully engulfed, is not a reliable methodology. Mouse microglia also differ from human microglia at the level of gene expression and activation states — a gap that further undermines the predictive value of these models.
A Phase 1 trial in healthy volunteers confirmed AL002 was safe and produced the expected spinal fluid signals. But healthy volunteers in their thirties tell you about safety, not whether a drug will slow neurodegeneration in elderly patients with established disease.
What the Trial Found: Active Drug, Passive Disease
The bottom line
Target engagement occurred — CSF sTREM2 fell and CSF osteopontin rose — but amyloid PET showed zero reduction, and nearly one in three treated patients developed amyloid-related imaging abnormalities (ARIA).
INVOKE-2 enrolled 381 people with early Alzheimer’s confirmed by amyloid biomarkers at 69 sites worldwide. Participants received AL002 at one of three doses or placebo by intravenous infusion every four weeks for up to two years.
The drug hit its pharmacological targets. sTREM2 fell in spinal fluid across all treated groups. Osteopontin — a protein linked to TREM2-driven microglial activation — rose. AL002 was reaching the brain and engaging TREM2. The biology was happening.
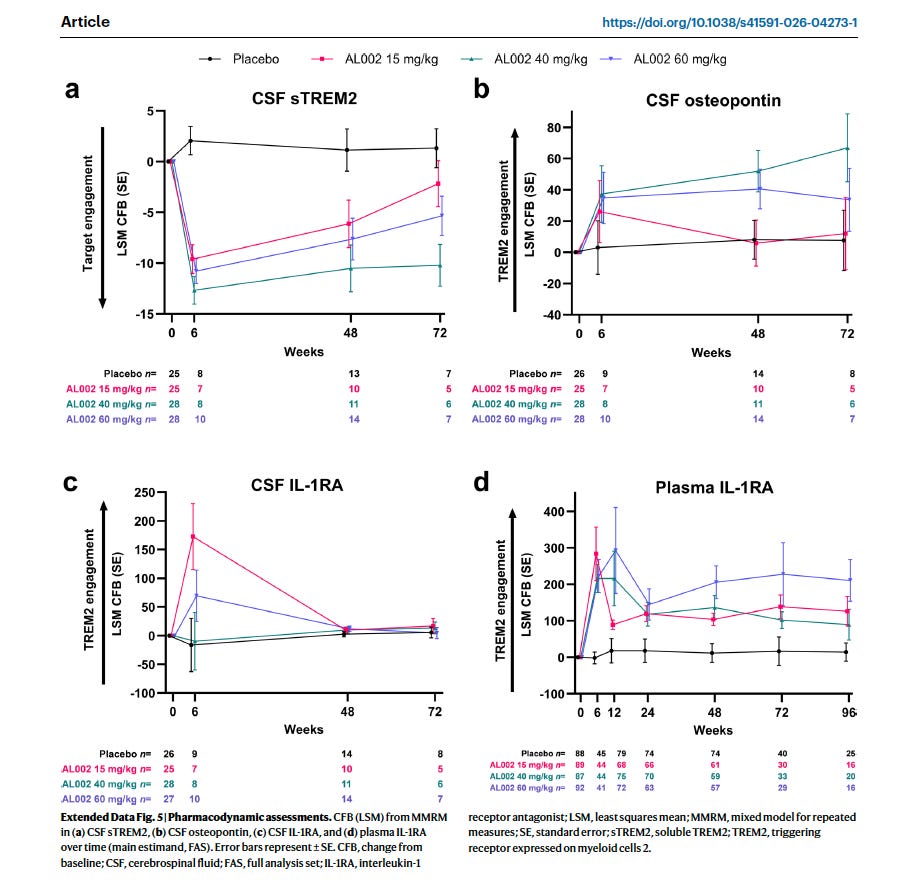
Figure 1: AL1002 engaged its target as shown with the drop in sTREM2, and raised Osteopontin, inflammatory markers. From Mummery et al, 2026
It just wasn’t helping. After 96 weeks, cognitive and functional decline was identical between treated and placebo groups across every measure tested — memory, orientation, daily function, composite scales. Blood and spinal fluid biomarkers of amyloid, tau, and neurodegeneration showed no difference. The disease progressed as if the drug were not there.
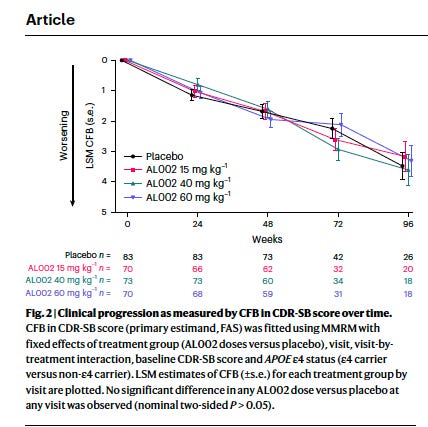
Fig 2: Clinical progression did not change after treatment, despite target engagement. From Mummery et al, 2026
What AL002 Actually Did to Microglia
Activated but not eating
The pharmacodynamic data tells a story, and it is not the story researchers hoped to tell. AL002 activated microglia — they moved toward plaques, enclosed nearby neurons, and protected them from the most acutely toxic fragments of amyloid. But critically, the drug did not activate the intracellular machinery that microglia need to actually engulf and digest an amyloid plaque. The downstream signaling cascade required — involving a pathway called PI3K and the physical restructuring of the cell’s internal skeleton — was not sufficiently engaged. The microglia arrived at the scene. They did not clean it up.
The result was what researchers now recognize as a Toxic Disease-Associated Microglia state — microglia that are biologically loud (secreting cytokines and inflammatory signals) but mechanically lazy (not phagocytosing the amyloid they were sent to clear). Sterile inflammation without productive action.
Osteopontin: a marker we misread
This reinterpretation fundamentally changes how we read one of the trial’s key biomarkers. CSF osteopontin (encoded by the SPP1 gene) was treated as a positive pharmacodynamic signal — evidence that TREM2 signaling had been activated and that microglia were responding. And in a narrow sense, it was. But osteopontin rising without a corresponding fall in amyloid is not a success marker. It is a failure marker.
High osteopontin drives microglia to activate the complement cascade — a set of immune proteins that tag structures for removal. The problem is that complement tags are not specific to amyloid. Microglia hypersensitized by complement activation mistake the molecular markers on healthy synapses — the connections between neurons — for debris, and prune them. The cleanup crew, activated but directionless, starts dismantling functional connections instead of toxic plaques. That pruning of healthy synapses is itself a driver of cognitive decline.
The lesson is subtle but important: moving toward a plaque is not the same as eating it. And eating the wrong thing is worse than not eating at all.
How the Drug Damaged Blood Vessels
The drug’s first encounter is not with microglia
AL002 is given by intravenous infusion — it enters the bloodstream. Before it ever reaches a microglial cell sitting next to an amyloid plaque deep in brain tissue, it must pass through the brain’s vasculature. Lining the outside of those blood vessels is a separate population of immune cells called perivascular macrophages (PVMs). PVMs also express high levels of TREM2, and AL002 binds them just as readily as it binds microglia.
When AL002 cross-links TREM2 receptors on a PVM, it triggers a pathway called Syk signaling — a molecular alarm that, in a PVM, is interpreted as a pathogen trying to breach the blood-brain barrier. The cell does not know it is responding to a drug. It responds as if an infection is trying to force its way into the brain. It begins preparing to demolish the vessel wall so that reinforcements — peripheral immune cells from the blood — can flood in.
In patients who also carry amyloid deposits within their blood vessel walls, a condition called cerebral amyloid angiopathy (CAA), the alarm is compounded. The PVM is simultaneously sensing the drug (interpreted as an invader) and vascular amyloid (interpreted as structural damage). The result is maximum alert: a large release of an enzyme called MMP-9 — molecular scissors that cut the structural collagen holding blood vessel walls together.
When the vessel wall fails
As MMP-9 dissolves the vessel wall, the blood-brain barrier fails. Fluid leaks from the bloodstream into surrounding brain tissue, causing swelling — this is ARIA-E (edema). Red blood cells also escape, releasing heme and toxic iron into brain tissue — this is ARIA-H (microhemorrhage). In APOE4 homozygotes, whose vessel walls are already structurally weakened and who carry the most CAA, the same mechanism produced the most severe outcomes: high rates of both ARIA types, and in some cases seizures, as heme disrupted the electrical signaling of neurons nearby.
This mechanism also explains why testing in standard APOE3 mouse models gave a false sense of safety. APOE3 blood vessels are structurally robust — the same MMP-9 release that ruptures weakened APOE4 vessels simply does not cause visible damage in a mouse with healthier vasculature. The risk was not detectable in the model used.
What This Means for the Next Generation of Microglial Drugs
New benchmarks — and a higher bar
AL002’s failure demands a reckoning with what we actually ask of drugs before they enter trials. Activation alone is no longer an acceptable endpoint. The field now needs to require three things that AL002 could not demonstrate: that a drug suppresses sterile inflammatory signaling rather than amplifying it; that it drives genuine amyloid phagocytosis — actual engulfment and lysosomal digestion — not just microglial recruitment; and that it is vascular-safe in APOE4-bearing models with pre-weakened vessel walls, not just in APOE3 mice whose stronger vessels will hide the damage.
A practical early-screening signal is emerging from this: before advancing any microglial drug, examine its transcriptomic fingerprint. If the SPP1 gene — which encodes osteopontin — dominates the signature without a parallel rise in amyloid-digestion and lysosomal genes, the drug is likely pushing microglia toward inflammatory noise rather than productive cleanup. That is the AL002 pattern. Catching it in the dish or the mouse, before a Phase 2 trial, could save years and patients.
Strategy 1: Refining TREM2 itself
Not everyone has abandoned TREM2 as a target — but the next wave of TREM2 drugs is being designed with AL002’s failures explicitly in mind. VHB937, currently in Phase 2 trials, takes a different approach: rather than activating and degrading the TREM2 receptor (as AL002 did), it stabilizes TREM2 at the cell surface, sustaining signaling without the receptor disappearing. Early data suggest it reduces pro-inflammatory biomarkers, which is the opposite of what AL002 accomplished. Whether stabilizing TREM2 drives more productive amyloid phagocytosis — rather than the inflammatory state AL002 induced — is the question the trial will answer.
A separate approach, VG-3927, recently acquired by Sanofi after positive Phase 1 data, takes a striking detour from the antibody format entirely. It is an oral small molecule TREM2 agonist. An oral drug distributes through the body differently from an intravenous antibody — potentially reaching microglia at lower, more sustained levels rather than flooding the system with a large pulse every four weeks. Whether that changes the vascular risk profile, and whether it drives phagocytosis or inflammation, are open questions for its upcoming Phase 2.
Strategy 2: Release the brakes instead of flooring the gas
Rather than forcing TREM2 to signal louder, a more physiological strategy is to remove the proteins that naturally suppress microglial phagocytosis. Three genetic targets identified in GWAS point this way.
SHIP1 (encoded by INPP5D) acts as a molecular brake downstream of TREM2 — it damps the PI3K signaling cascade that drives microglial engulfment of amyloid. Inhibiting SHIP1 removes that brake, enhancing amyloid uptake and lysosomal capacity in primary microglia without requiring TREM2 agonism. The catch, as described above, is that SHIP1 inhibition would also disinhibit perivascular macrophages, risking the same MMP-9-driven vascular damage seen with AL002. One proposed solution is combining SHIP1 inhibition with cPLA2 blockade — which would physically prevent PVMs from producing the enzyme that damages blood vessel walls, leaving microglia empowered to eat plaques while the vascular risk is contained.
PILRA is a second inhibitory receptor on microglia whose protective variant (G78R) is associated with reduced Alzheimer’s risk, particularly in APOE4 carriers. Recent work published in Science Translational Medicine showed that blocking PILRA with a high-affinity antibody rescued the metabolic and phagocytic deficits that APOE4 specifically causes in microglia — reducing amyloid pathology and restoring synaptic integrity in mouse models transplanted with human microglia. This makes PILRA especially interesting as a target in APOE4 carriers, potentially addressing a genotype that drove the most severe complications in INVOKE-2.
CD33 (Siglec-3) is a third inhibitory receptor whose loss-of-function variants are protective in Alzheimer’s GWAS. CD33 suppresses TREM2 signaling and blocks microglial phagocytosis partly through interaction with SHIP1. A CD33-blocking antibody (AL003) was discontinued in 2022, but the target remains biologically valid — and given what we now know about what AL002 lacked, a drug that disinhibits phagocytosis through CD33 blockade rather than activating inflammation through TREM2 agonism may deserve fresh attention.
Strategy 3: Dampen the inflammatory fire
A third approach asks a different question: rather than trying to redirect microglial activity toward phagocytosis, what if we simply reduce the inflammatory damage those microglia are causing? Two drugs in Phase 2 trials test this idea directly. XPro1595 is a selective TNFα inhibitor designed to block type 1 TNF receptors — the ones that drive neuroinflammation — while leaving type 2 receptors intact, which support myelin maintenance and protective immune functions. The goal is to cool the inflammatory microglial state without broadly suppressing the immune system. Canakinumab, an antibody against IL-1β, targets a specific inflammatory cytokine that activated microglia produce in abundance. Neither drug is designed to drive amyloid clearance directly — they aim to reduce the collateral damage that inflamed microglia cause while other mechanisms handle the plaques.
Strategy 4: Fix the digestive machinery
Even microglia that successfully engulf amyloid may fail to destroy it if the cellular machinery for digestion is impaired. This is an underappreciated dimension of the problem, and it points toward a fourth class of targets: the lysosomal system inside the cell that is supposed to break down ingested material. Progranulin, encoded by GRN, is a protein that supports lysosomal health in microglia. Loss-of-function mutations in GRN cause frontotemporal dementia, and progranulin deficiency impairs the microglial ability to clear amyloid even when phagocytosis is initiated. More broadly, activating TFEB — a master transcription factor that drives production of lysosomal enzymes — could theoretically turn phagocytically recruited microglia into effective digesters, completing the process that AL002 started but could not finish.
These are not fully formed clinical programs yet, but they represent the conceptual shift that INVOKE-2 has accelerated: from asking ‘did we activate microglia?’ to asking ‘did the microglia actually eat the right thing, and digest it?’
Take-Home Messages
Finding a risk gene is not a treatment roadmap. TREM2 is genetically validated and biologically important. But knowing that a gene matters is very different from knowing when, how, and in which patients to intervene on it.
Mouse models have limits that matter. The 5xFAD model produces aggressive, artificial disease in pre-symptomatic mice using a molecule that wasn’t AL002. This model is useful for certain readouts, but not sufficient. Preclinical AD testing remains a formidable task.
Engaging a target is not the same as helping a patient. AL002 hit TREM2, activated microglia, and reached the brain. None of that translated into clinical benefit. Amyloid clearance may prove to be a useful efficacy biomarker.
Microglia that move are not microglia that eat. AL002 drove recruitment without phagocytosis. The next generation of drugs must demonstrate actual amyloid digestion — not just inflammatory activation — before advancing to trials.
Rising osteopontin was not a success signal — it was a failure signal. High CSF osteopontin reflects inflammatory, synapse-pruning microglia, not productive amyloid clearance. Inflammation is a tough target and a complex readout.
The vascular damage had a specific mechanism. AL002 activated TREM2 on blood vessel-lining macrophages, not just neurons. Those cells released MMP-9, cut structural collagen, and damaged the blood-brain barrier — an effect invisible in standard mouse models but devastating in APOE4 patients.
Future microglial drugs need a vascular safety test. ARIA-like damage only appears in APOE4 models with pre-weakened vessels. Testing in APOE3 mice provides false reassurance. That benchmark must be built into preclinical programs.
The next drugs must prove they actually clean up, not just show up. The field is now pursuing four distinct strategies and fixing the cellular digestive machinery (lysosomal and progranulin approaches) is in the pipeline. We have reasons to be hopeful for a breakthrough.
Sources
Mummery CJ et al. The TREM2 agonistic antibody AL002 in early Alzheimer’s disease: a phase 2 randomized trial. Nature Medicine (2026). https://doi.org/10.1038/s41591-026-04273-1
Guerreiro R et al. TREM2 variants in Alzheimer’s disease. N Engl J Med 368, 117–127 (2013).
Jonsson T et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 368, 107–116 (2013).
Jain N et al. Chronic TREM2 activation exacerbates Aβ-associated tau seeding and spreading. J Exp Med 220, e20220654 (2023).
Etxeberria A et al. Neutral or detrimental effects of TREM2 agonist antibodies in preclinical models of Alzheimer’s disease and multiple sclerosis. J Neurosci 44, e2347232024 (2024).
Wang S et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J Exp Med 217, e20200785 (2020).
Zhong L et al. Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat Commun 10, 1365 (2019).
Edwin TH et al. A high cerebrospinal fluid soluble TREM2 level is associated with slow clinical progression of Alzheimer’s disease. Alzheimers Dement (Amst) 12, e12128 (2020).
Cummings J et al. Alzheimer’s disease drug development pipeline: 2025. Alzheimers Dement (N Y) (2025). https://pmc.ncbi.nlm.nih.gov/articles/PMC12131090/
Loss of PILRA promotes microglial immunometabolism to reduce amyloid pathology in cell and mouse models of Alzheimer’s disease. Science Translational Medicine (2025). https://doi.org/10.1126/scitranslmed.adw7428
VHB937 Phase 2 trial in Alzheimer’s disease — NeurologyLive (2025). https://www.neurologylive.com/view/new-phase-2-trial-test-trem2-stabilizing-agent-vhb937-alzheimer-disease
Vigil Neuroscience VG-3927 Phase 1 data (2025). https://www.globenewswire.com/news-release/2025/1/23/3014135/0/en/Vigil-Neuroscience-Reports-Positive-Data-from-its-Phase-1-Clinical-Trial-Evaluating-VG-3927-for-the-Potential-Treatment-of-Alzheimer-s-Disease.html
Sanofi acquires Vigil Neuroscience (2025). https://www.sanofi.com/en/media-room/press-releases/2025/2025-05-21-23-15-31-3086232
Optimization of SHIP1 inhibitors for the treatment of Alzheimer’s disease. PMC (2025). https://pmc.ncbi.nlm.nih.gov/articles/PMC11713407/
Thanks for another great post! My 30,000 ft, I-know-enough-to-be-dangerous view has always been that AD has its roots in the brain's immune system. It just makes too much sense. The answers are there and those early failed trials still gave us valuable data. At my age, my best offense is a great defense to mitigate risk, but I'm super excited for the treatment options that will be available for the next generations.
What did you make of Rudy Tanzi’s group’s recent work showing female immunosuppression benefitted cognition but to do it in males worsened cognition?