

When Your Brain's Immune System Burns Out: A New Way to Think About Alzheimer's Disease
What if Alzheimer's isn't just about the buildup of toxic proteins — but about the immune cells that were supposed to clean them up, and why they eventually stop doing their job?

There is a story we’ve been telling about Alzheimer’s disease for decades. Sticky plaques of amyloid-beta protein accumulate in the brain. Tangles of tau protein strangle neurons from the inside. The brain shrinks, synapses fail, memories dissolve. The story is true, as far as it goes. But it leaves out one of the most important characters: the immune system.
Over the past ten years, a new picture has emerged from genetics, from single-cell biology, and from some surprising places — the Bolivian Amazon, a Norwegian natural experiment on shingles, a ward of bladder cancer patients getting TB vaccines. The evidence is pointing toward a unified idea: that Alzheimer’s disease is, at least in part, a disease of immune exhaustion. The brain’s immune cells — microglia — become overwhelmed, worn out, and eventually unable to do the maintenance work that keeps Alzheimer’s pathology from taking hold. And some of our most powerful genetic risk factors, including the notorious APOE4 gene, may be accelerating exactly that process.
Understanding this reframes a lot of things: what our most recent drugs are actually doing, why certain vaccines seem to protect against dementia, and most importantly, what we should be developing next.
The Alzheimer’s Genome Is a Microglia Blueprint
To understand why the immune system matters so much in Alzheimer’s, start with genetics. Over the past fifteen years, genome-wide association studies (GWAS) — massive hunts through the DNA of hundreds of thousands of people for variants linked to disease — have uncovered more than 40 genetic locations associated with late-onset Alzheimer’s disease risk.
When researchers look at what those genes actually do, a striking pattern emerges. A large fraction of them are not expressed primarily in neurons. They are expressed in microglia — the resident immune cells of the brain. Genes like TREM2, CD33, BIN1, INPP5D, ABI3, CR1, and SPI1 all have primary functions in microglial biology: regulating how these cells respond to danger, how they engulf and degrade debris, how they signal to one another and to neurons.
The GWAS picture, in other words, tells us that the genetic architecture of Alzheimer’s is, to a remarkable degree, an immune architecture. Whatever is going wrong in the disease, the immune system is centrally implicated — not as a bystander, but as a principal actor.
TREM2 (Triggering Receptor Expressed on Myeloid Cells 2) deserves special mention. Rare variants in TREM2, particularly the R47H variant, confer a risk of late-onset Alzheimer’s roughly comparable to carrying one copy of APOE4. TREM2 is a receptor that sits on the surface of microglia and helps them sense and phagocytose — essentially, eat — cellular debris including amyloid plaques. When TREM2 is impaired, microglia lose their ability to effectively wrap around and dismantle plaques. They also struggle to survive in the inflammatory milieu of the Alzheimer’s brain. The result is a brain that is literally less capable of cleaning itself.
Microglia: The Brain’s Tireless Caretakers
To appreciate what goes wrong, it helps to appreciate what goes right — at least when things are working well.
Microglia make up roughly 10–15% of all brain cells. They are the central nervous system’s resident immune cells, derived from progenitor cells in the yolk sac during fetal development and maintained largely independently of the circulating bloodstream thereafter. They are, in the most literal sense, the brain’s dedicated immune workforce — present throughout life, patrolling constantly, responding to disturbances.
In a healthy brain, microglia perform ongoing surveillance. They extend fine processes that probe the local environment, sensing cellular damage, pathogen-associated signals, or the accumulation of abnormal proteins. When they detect a problem, they can migrate to the site, engulf and degrade the problematic material, and coordinate a local immune response to resolve the threat. They also play important roles in normal brain homeostasis — pruning synapses during development, supporting neuronal survival, and maintaining the extracellular environment.
In the context of Alzheimer’s disease, microglia are responsible for surveilling and clearing amyloid-beta plaques and tau tangles — the canonical hallmarks of the disease. When amyloid accumulates, microglia cluster around plaques in what is called a disease-associated microglia (DAM) state, upregulating genes involved in phagocytosis and inflammatory signaling. This response is protective, at least initially. It slows plaque expansion and contains the damage.
The problem is that this activated state has a cost. Sustained microglial activation is metabolically demanding and pro-inflammatory. If the stimulus — the accumulating amyloid, the chronic low-grade infection, the cellular stress — never resolves, the microglia cannot return to a resting homeostatic state. They are trapped in a cycle of activation without resolution.
When Immune Cells Run Out of Steam: Terminally Inflammatory Microglia
What does immune exhaustion look like in the brain? A landmark 2024 paper in Immunity by Millet, Ledo, and Tavazoie at Rockefeller University provides one of the most detailed answers yet (Millet et al., Immunity, 2024). Using single-cell RNA sequencing across an entire atlas of brain immune cells in Alzheimer’s model mice bearing different human APOE alleles, the researchers identified a distinct population of microglia they termed Terminally Inflammatory Microglia, or TIMs.
TIMs are characterized by simultaneous expression of inflammatory signaling markers and cellular stress markers. They are not simply “activated” microglia in the conventional sense — they appear to be something more advanced and more problematic: a population that has exhausted its adaptive capacity. Using trajectory analysis, the authors showed that TIMs represent a terminal state in microglial biology, arising from homeostatic microglia through acutely and chronically inflammatory intermediate states. Critically, once microglia reach the TIM state, it appears to be largely irreversible.
The frequency of TIMs was markedly elevated with age and with APOE4 genotype. In very old AD model mice, TIMs made up 45% of all microglia in APOE3 mice — but a striking 69% in APOE4 mice. This population was also detectable in human Alzheimer’s brains, with higher frequency in APOE4 carriers and at more advanced stages of disease (higher Braak score). Perhaps most damning: TIMs showed severely impaired capacity to phagocytose amyloid-beta. In direct ex vivo phagocytosis assays, TIMs were dramatically underrepresented among cells that successfully cleared amyloid. And in APOE4 mice, even TIMs that retained some phagocytic capacity were less effective than their counterparts in other genotypes.
The picture that emerges is stark. APOE4 does not merely accelerate amyloid production or impair its clearance through lipid metabolism. It also drives microglia faster toward a state of immune exhaustion — a state from which they cannot effectively do the work that might protect the brain.
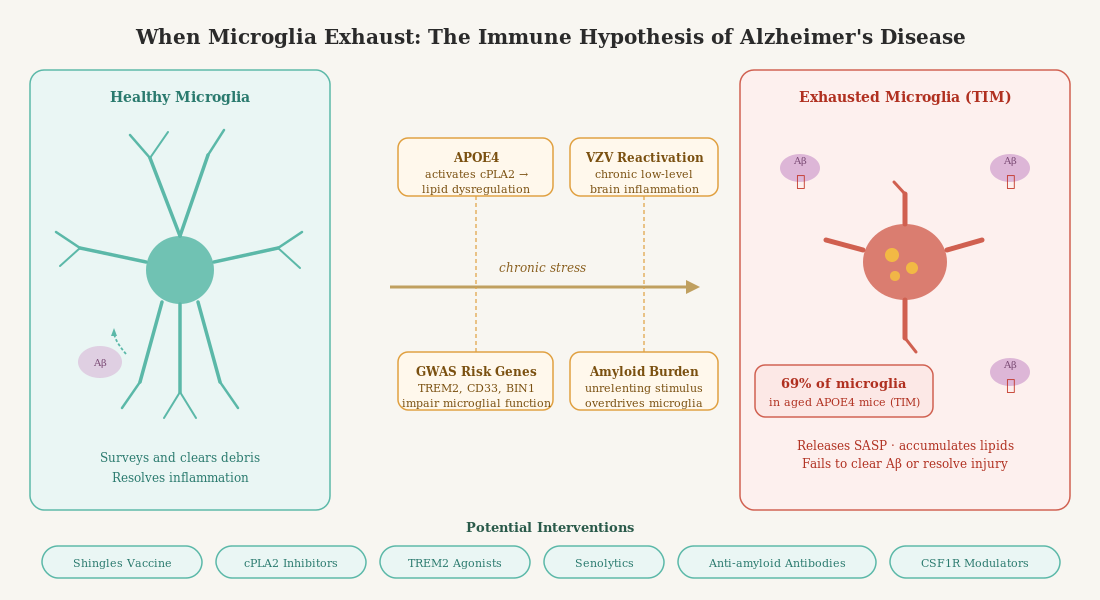
Illustration created using Claude.
APOE4: A Risk Gene That Made Evolutionary Sense
The APOE4 story has long been puzzling from an evolutionary perspective. If the allele is so harmful — raising lifetime Alzheimer’s risk by 3-4 fold, reducing longevity — why does it exist in 20–25% of most human populations? Why hasn’t natural selection eliminated it?
A remarkable 2017 study by Trumble, Stieglitz, Blackwell, and colleagues at Arizona State University and UC Santa Barbara helps answer this question (Trumble et al., FASEB Journal, 2017). They studied 372 members of the Tsimane people — Amazonian forager-horticulturalists in Bolivia who live without access to sanitation, clean water, or modern medicine. More than two-thirds of Tsimane adults carry active intestinal helminth (parasite) infections at any given time.
The finding was startling. In industrial populations, APOE4 reliably predicts cognitive decline. But among Tsimane adults with high parasite burden, the opposite was true: APOE4 carriers showed better cognitive performance than non-carriers, particularly on measures of fluid cognition. The higher the parasitic load (measured by eosinophil counts, a biomarker of infection), the more pronounced the cognitive advantage of carrying APOE4. In parallel, APOE4 carriers in this population had significantly lower eosinophil counts overall — suggesting the allele was also helping to actually clear parasitic infections.
The interpretation is that APOE4 evolved in an environment of heavy pathogen burden. Its effects on immune function — including its effects on cholesterol metabolism, lipid transport, and immune signaling — were adaptive when the immune system was constantly challenged by real parasitic and infectious threats. These are conditions that characterized the vast majority of human evolutionary history. What has changed is the environment. In modern industrialized populations with low pathogen burden, the immune-stimulating properties of APOE4 no longer have a useful target. Instead of keeping a useful immune response calibrated against real threats, APOE4 may push microglia toward a state of chronic, unresolved inflammation — the very condition that drives immune exhaustion.
This is the mismatch hypothesis applied to the immune system: APOE4 was built for a world of worms and bacteria, and in a world of processed food and antibiotics, it may be driving the brain’s immune system toward burnout.
Microglial Senescence: When Tiredness Becomes Permanent
Immune exhaustion and cellular senescence are related but distinct concepts, and both are increasingly implicated in Alzheimer’s disease.
Cellular senescence is a state in which cells permanently exit the cell cycle, lose normal function, and begin secreting a cocktail of pro-inflammatory molecules — a phenomenon known as the senescence-associated secretory phenotype (SASP). Senescent microglia have been identified in aging human and mouse brains, and their numbers increase dramatically with age and in the presence of AD pathology. They are characterized by dystrophic morphology (fragmented, bulbous processes rather than the fine ramified extensions of healthy microglia), impaired phagocytic capacity, and elevated expression of stress-response genes.
There is a vicious cycle at work here. Chronic microglial activation in response to accumulating amyloid and tau generates persistent inflammatory signals. These signals promote cellular stress and, eventually, senescence. Senescent microglia then cannot clear pathology — allowing it to accumulate further — and actively contribute to neuroinflammation through SASP. The TIM population described by Millet et al. appears to represent something analogous: a late-stage, functionally compromised microglial state that can no longer effectively contribute to amyloid clearance, and that may be enriched for senescent markers.
The key point is that this is not simply “inflammation” in the cartoon sense of the immune system being turned on. It is chronic, unresolved, dysregulated inflammation — a system that has lost the capacity to mount and then resolve an effective immune response. It is inflammation as dysfunction, not inflammation as defense.
The Shingles Vaccine: An Accidental Experiment in Immune Rescue
One of the most intriguing recent threads in Alzheimer’s prevention comes not from neuroscience labs, but from large population studies of vaccination.
In 2025, a landmark natural experiment published in Nature Medicine exploited a Welsh policy quirk: the live-attenuated shingles vaccine (Zostavax) was rolled out in age-cohort order, meaning that people born just before a certain date were eligible and people born just after were not. This created a near-randomized comparison. The result: vaccine eligibility was associated with a roughly 20% reduction in dementia incidence over the next several years — a striking effect size for a vaccine not designed to protect against dementia.
This finding was complemented by a 2026 study by Kim and Crimmins in the Journals of Gerontology, using data from the Health and Retirement Study (Kim & Crimmins, J Gerontol, 2026). Among nearly 4,000 older Americans, those who had received the shingles vaccine showed significantly more favorable biological aging profiles, including lower systemic inflammation, slower epigenetic aging (as measured by DNA methylation clocks), and lower transcriptomic aging scores. Crucially, the inflammation reduction was robust even after adjusting for socioeconomic factors, health behaviors, and multimorbidity. The effect was most pronounced in the years immediately following vaccination, consistent with the hypothesis that suppressing varicella-zoster virus (VZV) reactivation reduces chronic immune stimulation.
The mechanism, the researchers propose, is one of immune recalibration. The herpes zoster virus lies dormant in nerve tissue after childhood chickenpox infection, and it periodically reactivates — sometimes producing shingles, often subclinically. Each reactivation triggers an immune response, elevating inflammatory cytokines like IL-6, TNF-alpha, and CRP. Over decades, this chronic, low-level immune activation contributes to inflammaging — the background hum of systemic inflammation that accelerates biological aging and may progressively exhaust immune capacity.
By suppressing VZV reactivation, shingles vaccination may reduce this chronic immune burden. Microglia, relieved of one source of sustained peripheral immune activation, may be better positioned to maintain homeostasis and respond effectively to amyloid and tau. In essence, the vaccine may be protecting the brain by giving the immune system one fewer thing to fight.
Importantly, Kim and Crimmins also note that influenza and pneumococcal vaccines showed some similar — if weaker — benefits, particularly in the cardiovascular domain. This raises the question of whether reducing the burden of latent and recurrent infections more broadly might constitute a strategy for preserving immune capacity in aging. Whether BCG vaccination (the tuberculosis vaccine, which has broad non-specific immune-modulating effects and has been associated with roughly a 20–45% reduction in Alzheimer’s risk in some retrospective studies), herpes simplex vaccines (not yet approved), or earlier pneumococcal vaccination might offer parallel protections is not yet known, but the hypothesis is increasingly testable.
What Anti-Amyloid Antibodies Are Really Doing
The approval of aducanumab in 2021 and lecanemab in 2023 brought the first disease-modifying treatments for Alzheimer’s disease to the clinic. These are monoclonal antibodies designed to bind amyloid-beta and accelerate its removal from the brain. They work — lecanemab reduced the rate of clinical decline by about 27% in phase 3 trials. But they also come with a significant side effect: Amyloid-Related Imaging Abnormalities, or ARIA.
ARIA manifests as brain edema (ARIA-E) or microhemorrhages (ARIA-H) on MRI. In clinical trials, ARIA-E occurred in about 12–35% of patients depending on the antibody and APOE4 genotype, with APOE4 carriers at substantially higher risk. It is usually asymptomatic but can cause headache, confusion, and in rare cases, serious neurological events.
The mechanism of ARIA illuminates something important about what these antibodies are actually doing. Anti-amyloid antibodies opsonize amyloid aggregates — they tag them for destruction. Microglia express Fc receptors that recognize the antibody-tagged amyloid and engulf it. But amyloid also accumulates extensively in cerebral blood vessels (cerebral amyloid angiopathy), and when the antibody binds this vascular amyloid, it triggers an immune response at the vessel wall — complement activation, microglial Fc receptor-mediated phagocytosis, and the resulting inflammation increases vascular permeability, producing the edema and microbleeds of ARIA.
The Millet et al. study sheds light on what happens to microglia during aducanumab treatment. Acute treatment drove microglia toward disease-associated (DAM) states and effector-hi TIMs — an expanded immune response. Crucially, aducanumab and APOE4 genotype were both associated with a higher number and strength of predicted cell-cell interactions in the microglial compartment, suggesting that the drug is doing more than simply tagging amyloid for clearance. It is engaging and reshaping the brain’s immune landscape, including expanding adaptive immune cell interactions (particularly CD8 T cells) in an APOE4-dependent manner.
This is both promising and cautionary. It suggests that anti-amyloid antibodies may be exerting some of their benefits — and their risks — by broadly engaging microglial biology beyond amyloid clearance. Whether they can help restore a more homeostatic microglial state, or whether they risk further exhausting already-compromised TIM populations, remains an important open question. In APOE4 carriers, where TIMs are more abundant and more profoundly impaired, these dynamics may be particularly consequential.
The Future: Restoring Microglial Biology
If the central problem in Alzheimer’s — at least through the lens of APOE4 and many GWAS hits — is the progressive exhaustion and senescence of microglia, what can we do about it?
Several approaches are in active development:
TREM2 agonists. TREM2 is a critical survival and activation signal for microglia, and its expression is reduced in aging and in advanced disease. Agonistic antibodies that activate TREM2 signaling — including AL002 (tested in the INVOKE-2 phase 2 trial) and VHB937 (currently in phase 2) — aim to restore microglial function by boosting this pathway. The INVOKE-2 results were disappointing on primary endpoints, raising questions about timing: TREM2 agonism may need to be applied earlier in disease progression, before the TIM state becomes dominant.
Senolytics. Drugs that selectively eliminate senescent cells are a rapidly growing field. In preclinical models, clearing senescent microglia reduced neuroinflammation and improved cognitive outcomes. Clinical trials in Alzheimer’s are in early stages, but the rationale is compelling: if a proportion of microglia have become permanently dysfunctional, removing them might allow the remaining healthy population — or newly recruited monocyte-derived cells — to function more effectively.
CSF1R modulators. CSF1R (Colony Stimulating Factor 1 Receptor) governs microglial survival and proliferation. Transient pharmacological depletion of microglia followed by withdrawal of the drug allows repopulation from progenitor cells — essentially resetting the microglial compartment. This approach has shown promise in preclinical settings, though getting the timing and context right for human application is an active area of investigation.
Cell therapy. More ambitiously, some researchers have explored transplanting healthy microglia or microglial precursors to replace exhausted resident populations. A recent study demonstrated that systemic hematopoietic cell transplantation can restore microglial TREM2 function in mouse models.
Metabolic support. TIMs, as Millet et al. showed, are characterized by profound metabolic derangement — depleted glycolysis, pentose phosphate pathway activity, and TCA cycle function. Whether interventions that restore microglial metabolic fitness (through mTOR modulation, NAD+ precursors, or other approaches) could help prevent or reverse the TIM state is an exciting but still largely unexplored question.
cPLA2 inhibition. One particularly compelling metabolic-inflammatory target is calcium-dependent phospholipase A2 (cPLA2), an enzyme that sits at the intersection of lipid metabolism and immune activation. When microglia are chronically stimulated, cPLA2 is activated — either by oxidative stress, MAPK signaling, or elevated intracellular calcium — and begins liberating arachidonic acid (AA) from membrane phospholipids. AA is then metabolized by cyclooxygenases and lipoxygenases into a cascade of pro-inflammatory eicosanoids: prostaglandins, leukotrienes, and thromboxanes. In the short term, this is part of a normal immune response. But when cPLA2 stays chronically active, the sustained eicosanoid output generates reactive oxygen species, promotes lipid accumulation in lysosomes, activates NF-κB, and drives microglia toward the very senescent phenotype described above — a vicious cycle of inflammation and cellular damage that is difficult to escape. Critically, APOE4 specifically activates cPLA2 through MAPK p38 signaling, a finding confirmed in both animal models and postmortem human brain tissue (Wang et al., 2022; Ebright et al., 2022, reviewed in Hugo et al., Antioxid. Redox Signal., 2024). In AD mouse models, cPLA2 protein levels are elevated in astrocytes surrounding amyloid plaques, and cPLA2 knockout mice show rescued cognition on maze tasks. This makes cPLA2 inhibition an attractive strategy — not simply to reduce inflammation downstream, but to interrupt the lipid-driven senescence program at its source, potentially before irreversible TIM states are reached. The key practical challenge is developing inhibitors that are potent, selective for cPLA2 over related phospholipase isoforms, and capable of crossing the blood-brain barrier. Several lead compounds exist (including ASB14780 and the structurally diverse series emerging from virtual screening platforms), and the goal is not to abolish cPLA2 activity entirely — which would impair normal immune signaling — but to bring its chronic overactivation back into a physiological range. APOE4 carriers, who show measurably higher brain cPLA2 activity, may represent the population most likely to benefit; [18F]fluoro-AA PET imaging, which tracks arachidonic acid kinetics as a surrogate of brain cPLA2 activity, is being developed as a companion biomarker to identify and monitor this at-risk group.
Neither Good nor Bad: Reframing Inflammation
Throughout this discussion, you may have noticed that we keep running into a paradox: inflammation is implicated in Alzheimer’s disease, but so is immune exhaustion. How can both be true?
The answer is that the popular framing of inflammation as simply “good” or “bad” is not adequate to describe what is happening in the aging brain.
In the context of Alzheimer’s disease, we need good inflammation: the acute, targeted, resolving immune response that allows microglia to detect, engulf, and degrade amyloid and tau, then stand down. This is the physiological function the system was designed for. We also need the capacity to resolve inflammation — to return to homeostasis after a threat has been addressed.
What we do not want — and what appears to be accumulating in aging APOE4 brains — is chronic, unresolved inflammation: the persistent low-grade immune activation that exhausts microglial capacity, drives cells into senescent and terminally inflammatory states, and ultimately impairs the very clearance functions that were supposed to protect against pathology.
This is why the shingles vaccine finding is so conceptually important. The vaccine is not directly anti-inflammatory — in fact, it triggers a robust immune response. But by resolving a source of chronic latent viral pressure, it may restore the immune system’s capacity to function effectively: acute responses that resolve, rather than chronic smoldering that never does.
The same logic applies to APOE4. In an environment of high pathogen burden (the Tsimane Bolivians with their helminth infections), the allele’s immune-enhancing properties are channeled into useful, targeted immune work that keeps cognitive function intact. In a modern, low-pathogen environment, the same properties may tip the immune system toward chronic activation without resolution — driving microglia toward burnout.
And it is the same logic that should guide how we think about the new anti-amyloid antibodies. They are not simply “reducing inflammation” — they are restructuring the brain’s immune response, engaging microglia, and reshaping cell-cell interactions. Whether they do so in a way that ultimately restores or further compromises microglial resilience will likely depend on when in the disease course they are given, in whom, and at what dose.
Where This Leaves Us
We are at an early but genuinely exciting moment in Alzheimer’s research. The convergence of GWAS genetics, single-cell biology, evolutionary medicine, and epidemiology is drawing us toward a coherent picture of what goes wrong in the most common genetic form of Alzheimer’s disease: the progressive failure of the brain’s immune workforce.
The discovery of TIMs — exhausted-like microglia that accumulate with age and APOE4 burden and cannot effectively clear amyloid — provides a compelling cellular mechanism for much of what we have observed clinically. The finding that APOE4 functioned differently — indeed, protectively — in environments with high parasite burden puts this in evolutionary context. The association between shingles vaccination and slower biological aging suggests that the immune system’s capacity to protect the brain may be more modifiable than we thought.
The implications for therapy are profound. Rather than asking only “how do we clear amyloid?” we should also be asking: “how do we keep the immune cells that clear amyloid from burning out?” Those are different questions, and they may call for different — and complementary — answers.
A brain in which the right inflammation happens at the right time, resolves completely, and leaves microglia refreshed and ready for the next challenge: that is what we are trying to preserve. Not a brain with no inflammation. Not a brain with relentless, unresolved inflammation. But a brain whose immune system can do its job — and then rest.
This post draws on four key papers:
Millet, Ledo & Tavazoie. “An Exhausted-Like Microglial Population Accumulates in Aged and APOE4 Genotype Alzheimer’s Brains.” Immunity 57(1): 153–170, 2024.
Trumble et al. “Apolipoprotein E4 is associated with improved cognitive function in Amazonian forager-horticulturalists with a high parasite burden.” FASEB Journal 31: 1508–1515, 2017.
Kim & Crimmins. “Association between shingles vaccination and slower biological aging: evidence from a US population-based cohort study.” Journals of Gerontology, Series A, 81(3): glag008, 2026.
Hugo, Asante, Sadybekov, Katritch & Yassine. “Development of Calcium-Dependent Phospholipase A2 Inhibitors to Target Cellular Senescence and Oxidative Stress in Neurodegenerative Diseases.” Antioxid. Redox Signal., 2024. DOI: 10.1089/ars.2024.0794
Additional references:
Late-onset AD genetics implicates microglial pathways — Molecular Neurodegeneration, 2017.
Functional characterization of AD genetic variants in microglia — Nature Genetics, 2023.
Senescent microglial accumulation in AD — Neuron, 2024.
Varicella-zoster reactivation and dementia risk — Nature Medicine, 2025.
Vaccines and dementia — BCG and beyond — J Alzheimers Dis, 2024.
TREM2 and sTREM2: mechanisms to therapies — Molecular Neurodegeneration, 2025.
Aducanumab induces sustained microglial alterations — Journal of Experimental Medicine, 2024.
Claude Sonnet 4.6 (Anthropic) was used to help put this article together and create the illustration. AI is not used to generate content.
Thank you for this article, Hussein! I keep thinking about how anti-amyloid antibodies might actually be exhausting already-compromised microglia in APOE4 carriers rather than rescuing them. The timing question feels underappreciated in the clinical conversation.