

What the World's Oldest Person Teaches Us About Inflammation and Longevity
No single diet, supplement, or habit made her 117.

In January 2023, a woman living in a care home in Catalonia, Spain, became the oldest verified living person on Earth. She had been born in San Francisco in 1907, moved to Spain as a child, and lived through two world wars, a pandemic, the death of a son, and the steady rhythms of Mediterranean life — yogurt every morning, walks through the garden, books, piano, family, friends. She died in August 2024, at 117 years and 168 days.
What made her remarkable was not just how long she lived but how. Despite her extraordinary age, she never developed Alzheimer’s disease, never had cancer, and showed no cardiovascular disease. A team of researchers in Spain performed what may be the most comprehensive biological study ever conducted on a single human being — analyzing her genome, transcriptome, metabolome, proteome, gut microbiome, and epigenome, comparing results against multiple matched cohorts. They published their findings in October 2025 in Cell Reports Medicine.1 What emerged was not a simple formula for longevity. It was a portrait of a life in which many biological systems remained, against considerable odds, in functional equilibrium.
Aging and disease can be decoupled
The data revealed an immediate paradox. This woman showed clear molecular signatures of extreme old age: her telomeres — the protective caps at chromosome ends that shorten with each cell division — were the shortest ever recorded in a healthy person. Her blood showed clonal hematopoiesis, in which a mutated blood stem cell expands over decades and is associated with elevated risk of hematological malignancy and cardiovascular disease. Her immune cells included an expanded population of age-associated B cells, which accumulate with aging and are linked to pro-inflammatory and autoimmune activity.
And yet she had none of the diseases these markers are associated with. The paper’s most important conceptual contribution is demonstrating that molecular aging and age-associated disease are not synonymous. They can be decoupled. The hallmarks of aging — telomere attrition, clonal mutations, immune cell remodeling — can accumulate while the body maintains functional health, provided that something is holding the downstream pathological consequences at bay.
Across every layer of her biology, that something kept appearing.
Low inflammation as the common signal
Measured across blood proteins, lipid particles, metabolites, and gene expression, one feature of her biology was consistently preserved: remarkably low systemic inflammation.
The most direct evidence came from two composite markers, GlycA and GlycB, measured by proton nuclear magnetic resonance spectroscopy. These signals reflect the circulating concentration of acute-phase glycoproteins — including haptoglobin, α1-antitrypsin, and transferrin — that the liver releases in response to inflammatory stimuli. In short-term immune responses, this is adaptive. When chronically elevated even at low levels, it reflects what researchers call inflammaging: the persistent, low-grade, unresolved inflammation that accumulates with age and drives deterioration across organ systems.
In large prospective cohort studies — including analyses of over 250,000 individuals from the UK Biobank — elevated GlycA independently predicts cardiovascular mortality, all-cause mortality, and dementia, beyond traditional risk markers.2 Her GlycA and GlycB were both exceptionally low at age 116. Her acute-phase inflammatory response was minimal.
This is not a trivial finding. It is the biological output of an entire life. And when examined alongside how she actually lived, it begins to make sense.
Diet: pattern, not protocol
She followed a Mediterranean dietary pattern and consumed three yogurts daily for at least the final two decades of her life.
The researchers found her gut microbiome was dominated by Bifidobacterium, particularly the family Bifidobacteriaceae — a pattern more typical of younger individuals. Most older adults show progressive Bifidobacterium decline with age, replaced by more pro-inflammatory species. In her case, this was reversed, and the abundance of Bifidobacterium tracked closely with her low-inflammation metabolomic profile. Bifidobacterium produces short-chain fatty acids and conjugated linoleic acid, both of which dampen inflammatory signaling in the gut mucosa and systemically. The researchers drew a direct link between her microbial composition and her low GlycA and GlycB values.
The yogurt strains she consumed — Streptococcus thermophilus and Lactobacillus delbrueckii subsp. bulgaricus — are known to support Bifidobacterium growth. But the yogurt did not function in isolation. It was embedded in a broader Mediterranean pattern: olive oil, vegetables, legumes, fish, minimal ultra-processed food. The anti-inflammatory lipid profile she displayed — very low VLDL and triglycerides, high HDL, favorable lipoprotein particle size distribution — is consistent with decades of this dietary pattern, not with any single food or nutrient.
Movement: habitual, not structured
She did not follow a structured exercise program. She walked, gardened, played piano, and cared for her dogs — continuous, low-intensity, purposeful movement embedded in daily life.
The distinction between habitual movement and structured exercise is biologically meaningful. Evidence from large cohort studies and accelerometry data suggests that reducing prolonged sedentary time and increasing low-intensity habitual movement confer substantial cardiovascular and metabolic benefits, including reductions in circulating TNF-α and IL-6, improved insulin sensitivity, and myokine-mediated anti-inflammatory signaling — effects that do not require high exercise intensity and that accumulate with consistency over years rather than sessions.3
Her movement was also cognitively rich. Piano performance engages motor control, auditory processing, working memory, attention, and emotional regulation simultaneously. Gardening requires ongoing problem-solving, seasonal planning, and fine motor adaptation. This overlap between physical and cognitive engagement is unlikely to be incidental.
Sleep: glymphatic clearance and immune maintenance
The paper notes that she maintained good sleep habits throughout her life. The biological significance of this extends well beyond rest.
During slow-wave sleep, the glymphatic system — a network of perivascular channels that becomes dramatically more active during sleep — clears metabolic waste from the brain, including amyloid-beta and tau, the proteins that aggregate into Alzheimer’s pathology. Even a single night of sleep deprivation measurably elevates amyloid-beta in cerebrospinal fluid. Chronic sleep disruption accelerates epigenetic aging, raises inflammatory markers including GlycA, impairs immune consolidation, and substantially increases dementia risk. The 2024 Lancet Commission on dementia prevention identifies sleep-disordered conditions among its 14 modifiable risk factors, which together account for approximately 45% of dementia cases worldwide.4
Sleep is also when the immune system consolidates memory and regulates inflammatory tone. As we discussed in our previous post, VZV reactivation — a driver of chronic inflammaging — is itself promoted by sleep disruption, creating a bidirectional loop between poor sleep and elevated systemic inflammation.
Chronic stress and psychological resilience
Her personal history included significant adversity, including the death of a son. The authors note she maintained strong physical and mental health throughout, remaining socially engaged and purposeful.
Chronic psychological stress activates the hypothalamic-pituitary-adrenal axis, sustaining elevated cortisol that progressively dysregulates immune function. Prolonged cortisol elevation activates NF-κB — the central transcription factor driving inflammatory gene expression — in circulating immune cells, accelerates telomere attrition, promotes hippocampal atrophy via reduced neurogenesis, and advances epigenetic aging. In longitudinal studies, high perceived stress in midlife independently predicts dementia decades later.5 Psychological resilience, operationally defined as the capacity to maintain regulatory function under adversity, is associated with lower cortisol reactivity, preserved immune regulation, and slower biological aging by multiple epigenetic clock measures.
Her resilience — likely supported by the same social network, sense of purpose, and daily structure that characterized her life more broadly — appears to have attenuated this pathway. These are not independent factors. Chronic stress disrupts sleep. Poor sleep elevates cortisol and inflammatory markers. Both impair glymphatic clearance and immune resolution. The feedback between stress, sleep, and inflammation is one of the most important amplification loops in biological aging.
Social connection and its biological effects
She remained embedded in a social network — family, friends, and caregivers — throughout extreme old age, including contact with animals.
Social isolation activates the same hypothalamic-pituitary-adrenal stress axis as physical threat, raising circulating inflammatory cytokines, accelerating telomere shortening, and predicting faster epigenetic aging. A major meta-analysis found loneliness associated with a 26% increase in all-cause mortality.6 The Lancet Commission identifies social isolation as a modifiable dementia risk factor. Mechanistically, perceived social connection downregulates NF-κB activity in immune cells, reduces cortisol, and promotes oxytocin signaling, which has direct anti-inflammatory effects. The biology does not appear to respond primarily to physical proximity but to the subjective experience of meaningful, reciprocal connection.
Cognitive engagement and reserve
She read books and played piano into her final years, and tended a garden that required continuous adaptation and problem-solving.
Formal education — measured by years of schooling — is identified by the Lancet Commission as the single largest modifiable dementia risk factor. The operative mechanism, however, is cognitive reserve: the brain’s accumulated capacity to sustain function in the presence of neuropathological damage. A brain with more synaptic redundancy and practiced cognitive flexibility can absorb more amyloid deposition, vascular injury, and neuronal loss before clinical symptoms appear. Post-mortem studies consistently show that individuals with high cognitive reserve may carry full Alzheimer’s pathology without clinical diagnosis.
Cognitive reserve is not fixed by formal schooling. It accrues across the lifespan through any genuinely demanding mental engagement. Piano performance is among the most neurologically complex activities available, requiring concurrent engagement of motor, auditory, mnemonic, attentional, and emotional systems. Her sustained engagement with cognitively demanding activities well past conventional retirement age likely continued building and maintaining the neural redundancy that may have protected her from symptomatic neurodegeneration.
A lifetime of infections, resolved
Her immune profile bore the signature of long experience. T cells were dominated by effector and memory subtypes. Immunoglobulin G levels — particularly IGHG2 and IGHG4 — were elevated, consistent with efficient and mature humoral memory. This immune phenotype reflects an organism that has encountered many pathogens, resolved the encounters, and consolidated the memory.
This is distinct from immune exhaustion, which we described in our previous post on microglial biology and Alzheimer’s disease: chronic, unresolved immune stimulation — from VZV reactivation, persistent amyloid burden, or other sources — drives immune cells toward a dysfunctional, pro-inflammatory, senescent state. Her immune system appeared experienced rather than exhausted: it had fought and cleared, repeatedly, across a century. This is consistent with the evolutionary framing introduced in our previous post — that APOE4 confers protection in high-pathogen environments by enhancing immune responsiveness, and becomes a liability in low-pathogen modern environments where the same responsiveness generates unresolved chronic inflammation.
She did not carry APOE4. Her APOE genotype was at the protective end of the spectrum, which almost certainly mattered for her lipid metabolism and neurological resilience.
Genetics helped, but gene-environment interaction was the mechanism
The researchers identified seven rare homozygous variants in her genome absent from European control populations, and protective configurations at APOE, FOXO3A, and genes involved in immune regulation, mitochondrial oxidative phosphorylation, cardiovascular function, and DNA repair.
Her genome was unusually favorable. But the paper is explicit: no single genetic variant explains her longevity. The identified variants span disparate biological processes — immune surveillance, energy metabolism, cardiovascular resilience, neuroprotection — and none appears individually sufficient. All appear to have operated in concert with each other and with her environment. MAP4K3, a longevity-regulating gene whose variant she carried, regulates lifespan in C. elegans through pathways that are sensitive to diet, stress, and metabolic state. The gene does not act independently of the environment in which it is expressed.
Gene-environment interaction is a core principle of quantitative genetics. The same variant that is protective in one environment may be neutral or deleterious in another. Her protective genome was expressed through — and likely required — the environmental context of her life: diet, physical activity, sleep, stress management, social engagement, and a century of immune experience.
The epigenetic clocks: an integrated readout
When the researchers estimated her biological age using six different epigenetic clocks — algorithms that infer biological age from DNA methylation patterns — every clock placed her substantially below her chronological age of 116. The discrepancy ranged from approximately 10 to 27 years across methods and tissues. Using the rDNAm clock, her biological age deceleration was 23.17 years.
Epigenetic clocks are accelerated by chronic inflammation, sleep disruption, psychological stress, social isolation, sedentary behavior, poor diet, and chronic unresolved infection — the same factors addressed throughout this post. Her 23-year deceleration represents the integrated biological output of a life in which these inputs were consistently attenuated. It is not attributable to any single variable. It reflects the cumulative effect of many factors operating in concert over a very long time.
What we do not know
Before drawing conclusions, it is important to be direct about the limits of this evidence.
This is a study of a single individual. No causal inferences can be drawn from an N of 1. We cannot determine whether her lifestyle choices produced her low inflammatory state, or whether her genetic endowment independently determined that state and also happened to promote the behaviors we observed — an instance of reverse causation that cannot be resolved in a cross-sectional study of one person.
We do not know the relative contribution of each factor discussed here. The multiomics data reveal associations between her biological profile and various features of her life and genome; they cannot quantify how much each factor mattered. The gut microbiome findings, for instance, are from a single time point late in life. We do not know whether her Bifidobacterium dominance was lifelong, whether it was a cause or consequence of her metabolic health, or whether it would have the same implications in a genetically different individual.
Epigenetic clocks, while powerful population-level tools, have been trained primarily on datasets that do not include many individuals past age 100. Their accuracy and interpretation at extreme ages remains uncertain. The variability and predictive power of these clocks are uncertain.
It is also worth noting that supercentenarians are not a homogeneous group. Other individuals who have lived to comparable ages have smoked, consumed alcohol heavily, and been largely sedentary. Extreme longevity likely has multiple biological routes, and the profile described here may represent one path among several. Survivorship bias is a genuine concern: we study those who reached extreme age, but we cannot easily study the many individuals who adopted similar lifestyles and did not.
Finally, most of the lifestyle factors described — diet, exercise, sleep, social connection — have been studied in relatively short-term randomized trials or observational studies spanning years to decades. Whether the effect sizes from those studies compound as expected over a full human lifespan is not established.
What the paper offers is not a proof of mechanism or recommendations for certain tests. It is a richly detailed portrait of one person whose biology was measurably younger than her age, in which low systemic inflammation emerges as the most consistent cross-cutting feature. That observation is worth taking seriously, even in the absence of causal certainty.
The sum is greater than the parts
What is most striking about this biological portrait is its refusal to yield a single active ingredient.
Not the yogurt. Not the Mediterranean diet. Not the walking. Not the piano. Not the protective genome. Not the Bifidobacterium. Not the good sleep. Not the family connections. No single factor, isolated, accounts for 117 years of health. The biology supports a systems view: these factors are mutually reinforcing, operating through shared mechanisms — principally, the regulation of chronic inflammation — and their combined effect is greater than any individual contribution.
A good high fiber fermented diet with an active lifestyle supports a healthy microbiome, which reduces systemic inflammation, which preserves epigenetic stability, which maintains mitochondrial function, which supports immune competence, which allows infections to resolve rather than persist, which protects sleep quality, which lowers cortisol, which further attenuates inflammation. These are not parallel pathways. They are a network. Single interventions improve one node in the network; a lifetime of consistent, modest, mutually reinforcing inputs keeps the network itself functional.
She did not optimize. She lived — with consistency, engagement, connection, and purpose — and the biology reflects it.
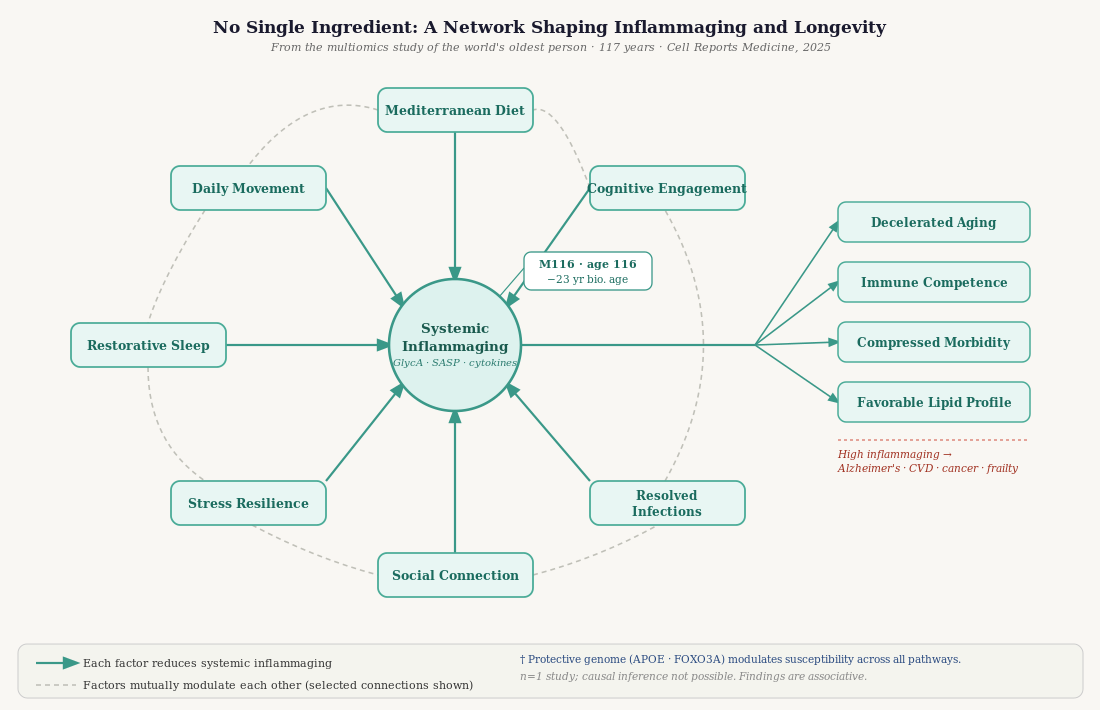
Illustration made using Claude
Take-home messages
There is no single intervention that confers longevity or health span. The most comprehensive biological study of an extreme long-liver found a confluence of genetic, dietary, microbial, immunological, and behavioral factors. The evidence does not support a protocol-based view of aging.
Inflammation is the final common pathway. Poor sleep, chronic stress, social isolation, physical inactivity, unresolved infection, and poor diet each generate chronic low-grade inflammation through overlapping mechanisms. Reducing the sources of that inflammation — rather than pharmacologically suppressing the immune response — is the central challenge of healthy aging.
Acute, resolving inflammation is not the problem — chronic, unresolved inflammation is. The immune system requires activation. Acute infections that clear, physical effort that triggers repair, and transient physiological stress that drives adaptation are all beneficial. The harmful signal is the one that never resolves.
Diet operates as a pattern over decades, not as an ingredient. The gut microbiome responds to the cumulative input of dietary diversity, fermented foods, fiber, and reduced ultra-processed intake. The evidence does not support supplementing individual components as a substitute for the overall pattern.
Habitual movement — independent of structured exercise — has measurable anti-inflammatory effects. Reducing prolonged sedentary time and sustaining low-intensity daily movement consistently improves metabolic and inflammatory biomarkers. Duration and consistency appear to matter more than intensity for most of the outcomes relevant to aging.
Sleep quality and duration affect amyloid clearance, immune regulation, and epigenetic aging. These are not independent consequences of aging — they are modifiable inputs that feed back into the aging process itself.
Chronic stress activates inflammatory signaling and accelerates biological aging by measurable epigenetic and telomeric markers. Psychological resilience and a sense of sustained purpose appear to attenuate this pathway.
Social connection downregulates NF-κB-mediated inflammatory signaling and is independently associated with dementia risk, cardiovascular outcomes, and mortality. The effect appears to be mediated by perceived meaningful connection rather than physical proximity alone.
Cognitive reserve — built by sustained engagement with mentally demanding activities — provides measurable protection against the clinical expression of neurodegeneration, even in the presence of pathological burden. It is not fixed by formal education and accumulates throughout the lifespan.
Genetics shapes vulnerability and resilience but does not determine outcomes independently of environment. Gene-environment interactions, particularly for variants like APOE4, mean that lifestyle factors may matter more — not less — for individuals with elevated genetic risk. The environment is where the greatest modifiable leverage lies.
The goal is the compression of morbidity — maintaining biological function close to the end of life — not the maximization of lifespan per se. She spent her final months managing bronchiectasis and osteoarthritis but reached that point without Alzheimer’s disease, cancer, or cardiovascular disease. That trajectory, more than the final number, is what the evidence points toward.
This post draws on: Santos-Pujol et al., “The multiomics blueprint of the individual with the most extreme lifespan,” Cell Reports Medicine 6, 102368 (2025); the Lancet Commission on Dementia Prevention, Intervention, and Care (2024 update); and concepts from our previous post on immune exhaustion and Alzheimer’s disease, including Kim & Crimmins (J Gerontol, 2026) and Millet et al. (Immunity, 2024). The post was constructed using Claude, Sonnet 4.6.
Footnotes
Santos-Pujol E et al. The multiomics blueprint of the individual with the most extreme lifespan. Cell Rep Med. 2025;6:102368. ↩
Wang Z et al. Plasma metabolomic profiles associated with mortality and longevity in a prospective analysis of 13,512 individuals. Nat Commun. 2023;14:5744.
Church S et al. Glycoprotein acetyls as a novel inflammatory biomarker of early cardiovascular risk. J Am Heart Assoc. 2022;11:e024380. ↩
Ungvari Z et al. The multifaceted benefits of walking for healthy aging. Geroscience. 2023;45:3211–3239. ↩
Livingston G et al. Dementia prevention, intervention, and care: 2024 report of the Lancet standing Commission. Lancet. 2024. ↩
Johansson L et al. Midlife psychological stress and risk of dementia: a 35-year longitudinal population study. Brain. 2010;133(8):2217–2224. ↩
Holt-Lunstad J et al. Loneliness and social isolation as risk factors for mortality. Perspect Psychol Sci. 2015;10(2):227–237. ↩