

To Statin or Not To Statin for Alzheimer's?
Why the best case for statins in APOE4 is vascular, not “Alzheimer’s treatment”

A 65-year-old man came in for a brain health consult. He is lean, exercises at least five days per week, and describes a diet most clinicians would call heart healthy. His mother developed dementia in her late 70s. Recently, he learned he carries two copies of APOE ε4 (APOE4/4).
What brought him in was a lab value that did not fit his lifestyle: LDL cholesterol 175 mg/dL. He had read conflicting messages, including claims that statins might worsen memory. He wanted to understand whether he should do something about his LDL, and how to think about that decision in the context of APOE4/4 and dementia risk.
So the question became:
If an APOE4/4 carrier has elevated LDL but no dementia, does lowering LDL with a statin meaningfully help brain health, and is there evidence that statins harm cognition?
This post is not medical advice. It is a way to read the evidence with the patient’s real-world decision in mind.
1) APOE4, high cholesterol, and ischemic heart disease: the “why” behind the lab
APOE is not just an “Alzheimer’s gene.” It is also a lipid transport gene. One practical consequence is that APOE4, especially APOE4/4, shifts lipid handling in ways that can raise LDL, even in people who eat well and exercise consistently.
Mechanistically, APOE sits on lipoprotein particles and helps them interact with hepatic receptors involved in clearance. In a randomized human study using a surface plasmon resonance approach plus hepatocyte uptake assays led by Anne Marie Mihihane and her team, APOE4 carriers showed altered lipoprotein–LDL receptor binding and downstream handling consistent with a propensity toward higher LDL under certain dietary fat conditions. This is one concrete example of how genotype can constrain physiology even when behavior is optimal.
The next step in the logic is vascular risk. Large population data support that APOE genotypes associate not only with dementia risk but also with ischemic vascular disease. For example, in UK Biobank (about 391,992 white British participants), APOE genotypes were associated with ischemic heart disease and relevant vascular risk factors, with effects that vary by genotype, age, and sex.
That matters for brain health because vascular injury, clinical stroke and “silent” small vessel disease, can lower cognitive reserve and amplify neurodegenerative trajectories. If someone is APOE4/4 and carrying a high LDL burden over time, it is reasonable to ask whether LDL lowering could reduce downstream vascular contributions to cognitive risk, even if it does not directly target Alzheimer pathology.
This is the first frame shift for the patient: his LDL cholesterol level may be partially genetic, and the brain relevance may be vascular as much as, or more than, direct Alzheimer prevention.
2) Genetic epidemiology signals: direction-of-effect, not proof
When randomized prevention trials are difficult, researchers often turn to genetic epidemiology to test whether a risk factor plausibly sits upstream of disease.
One example used ADNI data (N = 1,534 across cognitively normal, early MCI, late MCI, and AD) and performed a Mendelian randomization (MR) analysis using APOE as a genetic instrument to examine whether higher total cholesterol might contribute to risk along the clinical spectrum. They reported that higher total cholesterol was associated with higher odds of MCI and AD diagnoses in APOE4-positive versus APOE3 carriers in their MR framework.
How should we treat this?
It is not a statin trial.
MR is best read as a direction-of-effect argument: lipid biology is plausibly upstream of risk, particularly in APOE4.
APOE is biologically pleiotropic, so MR assumptions can be strained. The takeaway is plausibility, not certainty.
Next comes the observational pharmaco-epidemiology signal (association, not proof): In the Chicago Health and Aging Project (CHAP; N = 4,807), statin initiation was associated with a lower hazard of incident clinical Alzheimer’s disease overall, and the association appeared stronger in APOE ε4 carriers (HR about 0.60 in ε4 carriers vs about 0.96 in non-carriers; interaction reported).
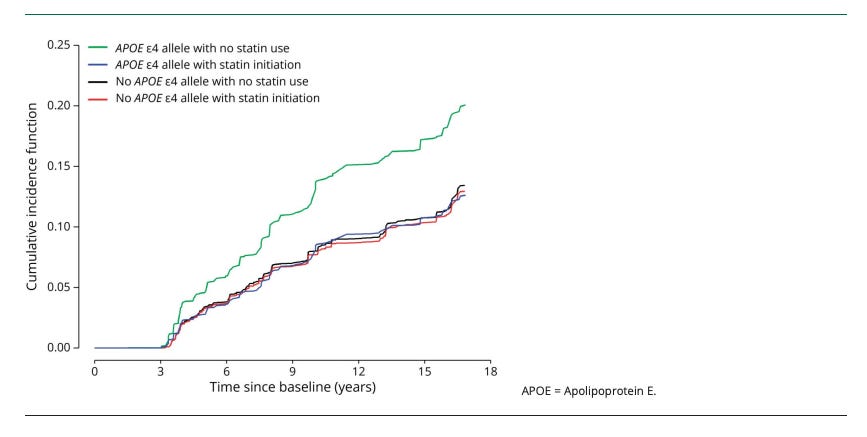
The figure above shows the covariate-adjusted cumulative incidence of AD Dementia, by statin initiation and the APOE e4 Allele
This type of result motivates clinicians and patients while also raising methodological caution, because healthy-user bias, confounding by indication, and time-varying treatment patterns are hard to fully eliminate.
So the honest interpretation is: there are credible signals suggesting subgroup benefit (notably in APOE4), but they are not definitive.
3) Why so many clinical trials are null, and why that may not settle prevention
If you only looked at randomized trials in people with established Alzheimer’s disease, you might walk away thinking the case is closed: statins do not help.
Two large symptomatic AD trials illustrate the point:
LEADe randomized 640 patients with mild-to-moderate AD to atorvastatin 80 mg/day vs placebo for 72 weeks and found no significant benefit on cognition (ADAS-Cog) or global function (ADCS-CGIC).
A separate randomized trial in 406 patients with mild-to-moderate AD using simvastatin vs placebo similarly reported no meaningful differences in cognitive or functional decline over 18 months.
These are important results, but they answer a treatment question (does this help after diagnosis) more than a prevention question (does decades of lower LDL reduce the probability of reaching symptomatic disease).
Why prevention is hard to prove in standard randomized clinical trials:
Dementia unfolds over years to decades, so trials need long follow-up and large samples.
People may cross over: they start and stop statins, and the cognitive risk benefit may change if the change diets, change blood pressure meds.
Cognitive endpoints can be relatively insensitive over short windows, especially in healthy or mildly impaired individuals.
If a therapy primarily reduces vascular events, the cognitive signal may be indirect and diluted, especially if neurodegeneration is already advanced.
So the null symptomatic trials are informative, but they do not fully answer what our patient cares about: risk modification earlier in life. One important finding from these trials is that even in patients with AD, statins do not worsen memory (more on that below). Newer trials for brain outcomes using statins should consider more brain biomarkers than cognition during the predementia phases, and especially those that reflect the effects of statins on the brain.
4) Debunking the idea that statins “worsen memory”
Patients commonly report cognitive symptoms while taking statins, but the key scientific question is whether statins cause clinically meaningful cognitive decline in controlled data.
A useful anchor here is that large randomized trials that included cognitive testing have generally not shown a consistent cognitive harm signal. For example, in PROSPER (5,804 participants aged 70 to 82 randomized to pravastatin vs placebo), detailed cognitive outcomes were assessed repeatedly and did not support a clinically meaningful adverse effect on cognition attributable to pravastatin.
A practical way to say this in clinic is: the best available evidence does not support the claim that statins generally worsen memory. If a specific patient experiences cognitive symptoms temporally associated with a medication, that should be evaluated carefully, but it should not be assumed to be the expected or typical effect.
5) Lipophilic and hydrophilic statins: why the distinction persists
Statins differ in chemical properties that influence distribution. Lipophilic statins are often discussed as having greater potential CNS penetration, while hydrophilic statins are generally more hepatoselective and do not enter the brain.
Does this translate into meaningfully different dementia outcomes? The evidence is not definitive, but large observational datasets have explored the question.
One widely cited example is a retrospective Medicare cohort of 694,672 beneficiaries that compared dementia risk across combinations of statins and antihypertensive classes. The authors reported that combinations involving pravastatin or rosuvastatin (hydrophilic statins) with renin-angiotensin system acting antihypertensives, particularly ARBs, were associated with lower dementia risk compared with some other combinations.
This is not causal proof, but it supports a reasonable scientific posture: drug class and combinations are plausible modifiers, and the question deserves prospective testing.
6) Uncertainties that remain, especially once Alzheimer’s disease is diagnosed
Two ideas should be kept separate:
Risk modification: lowering LDL and reducing vascular events, which plausibly supports brain resilience.
Disease modification: changing core Alzheimer’s pathology trajectories (amyloid, tau, neurodegeneration).
The large symptomatic AD trials above were largely null on cognition, which argues against presenting statins as established disease-modifying Alzheimer therapies once dementia is present.
For an APOE4/4 carrier without dementia, the strongest rationale remains vascular risk reduction. Whether LDL-lowering meaningfully changes dementia incidence is still uncertain, in part because it is difficult to run long, well-powered prevention trials for a peripheral target. The implication of this analysis makes us question APOE4 carriers with low LDL levels. Would they benefit from statin treatment if they had a lower cardiovascular risk profile?
Back to the case: risk-benefit framing
For this 65-year-old APOE4/4 man with LDL 175 mg/dL and no dementia, the most evidence-based framing is vascular.
APOE4/4 is associated with higher ischemic heart disease risk in large population data.
Elevated LDL is a modifiable vascular risk factor, and vascular events are relevant to brain health.
The best controlled evidence does not support a general claim that statins worsen cognition.
So, should he take a statin?
What can be said responsibly, based on the evidence reviewed, is that an LDL of 175 mg/dL at age 65 commonly triggers a clinician-patient discussion about lipid-lowering for vascular risk reduction. APOE4/4 and family history make the prevention context more salient, but they do not turn statins into a proven dementia-prevention drug.
The decision belongs in a discussion with his clinician that incorporates his overall cardiovascular risk profile, family history, preferences, and tolerance, while keeping expectations calibrated: the clearest benefit is vascular, the long-term dementia prevention-related benefit is plausible but not proven.
Take-home messages
APOE4/4 can raise LDL through lipid handling biology, even when lifestyle is excellent.
APOE genotypes, including ε4ε4, are associated with ischemic heart disease risk in large population data.
Genetic and observational studies provide plausible signals that lipid biology and statin exposure may matter more in APOE4, but they are not definitive prevention proof.
Large symptomatic Alzheimer’s trials of statins have been largely null, which does not settle prevention questions.
The best controlled evidence does not support the claim that statins generally worsen memory.
Whether to start a statin should be decided in discussion with a clinician, weighing vascular risk, LDL level, patient values, and tolerance, without assuming statins are a proven dementia-prevention therapy.
The benefit of statin initiation in those with low cardiovascular risk or low LDL levels for AD is uncertain.
This is a really useful question to put in public, because “statins + Alzheimer’s” is one of those topics where people get pulled into extremes: either statins melt your brain or statins prevent dementia for everyone. The truth is more nuanced, and your framing helps.
A few things I think readers benefit from holding at the same time:
Statins were never designed as “Alzheimer’s drugs,” but they are one of the cleanest levers we have for reducing ASCVD and ischemic stroke risk. And since vascular injury and neurodegeneration often travel together, improving vascular risk profiles is one of the most plausible ways to reduce some dementia risk at the population level. That’s part of why observational studies so often find an association between statin use and lower dementia incidence, especially in people with higher baseline cardiometabolic risk. 
At the same time, association isn’t causation. “Statin users have fewer dementia diagnoses” can reflect confounding (healthcare access, adherence behaviors, baseline risk differences), and we should be careful not to oversell what the data can support. I appreciated that you’re approaching this as a decision under uncertainty rather than a morality play. 
And finally: the fear that statins “cause dementia” has not held up well in the best available evidence. Some people do report short-term cognitive symptoms, but broad claims of increased dementia risk aren’t supported, and major research/clinical orgs continue to emphasize that the bigger brain risk signal is often untreated vascular risk over decades. 
The way I’d translate this into a practical takeaway for readers: if you’re considering a statin, it’s rarely a “yes/no for Alzheimer’s.” It’s usually a vascular risk management decision (LDL, ApoB, diabetes status, blood pressure, smoking, family history, CAC score when appropriate), with brain health as a meaningful downstream beneficiary if it helps keep the vascular system intact.
If you write a follow-up, I’d love to see how you’d counsel the tricky edge cases, such as very old adults, people with low baseline LDL, or those with prior intolerance, because that’s where individualized risk/benefit discussions really matter.
Hi! I posted about a large study on statins vs Alzheimer's here: https://substack.com/@drdorota/note/p-194206704?r=5usivi&utm_medium=ios&utm_source=notes-share-action