

The Vascular Side of Alzheimer's: A New Hypothesis for Why the Brain Bleeds
A fresh Perspective on vascular inflammation in AD

Alzheimer’s disease is not just a disease of plaques in the brain. It is also, and perhaps equally, a disease of blood vessels. In many patients — especially those carrying the high-risk APOE4 gene — amyloid accumulates inside the walls of the brain’s blood vessels, not just between neurons. This condition, called cerebral amyloid angiopathy (CAA), damages the vessels, triggers inflammation, and causes the blood-brain barrier to leak. It contributes to cognitive decline in its own right, and it makes the brain far more vulnerable to hemorrhage and swelling.
This vascular dimension of Alzheimer’s has long been underappreciated and undertreated. We do not yet have a drug that directly targets the vascular inflammation driving it. Ambreen Kanwal and Bilal Kerman, leading this work at USC, set out to ask whether a specific enzyme — called cPLA2 — might be that target. Their perspective article, just published in Alzheimer’s & Dementia, argues that cPLA2 sits at the intersection of amyloid burden, APOE4-driven lipid dysregulation, and blood-brain barrier breakdown, and that it may be a key upstream driver of vascular injury in Alzheimer’s disease. This post walks through the biology behind that hypothesis, the evidence supporting it, and what would be needed to test it in patients.
Cerebral Amyloid Angiopathy (CAA): Amyloid in the Wrong Place
Most people understand that Alzheimer’s disease involves amyloid plaques building up between brain cells. What is less appreciated is that amyloid also accumulates in the walls of the brain’s blood vessels — the arteries, arterioles, and capillaries that supply the brain with oxygen and nutrients. This condition is called cerebral amyloid angiopathy, or CAA, and it is far more common than most people realize.
Neuropathology studies show that CAA is present in the brain tissue of nearly all patients with Alzheimer’s disease, especially in older patients and in those who carry the APOE4 genetic variant. Even in cognitively normal elderly adults, CAA can be found in 20 to 40 percent of brains examined at autopsy. It is, in this sense, a near-universal companion of aging — but in Alzheimer’s, it is particularly prominent and particularly consequential.
CAA is not one uniform condition. Researchers distinguish between two main subtypes based on which vessels are affected. Type 1 CAA involves amyloid deposits in the basement membranes of the tiny capillaries — the smallest blood vessels deep in the brain tissue. Type 2 CAA, which is more common in older, cognitively normal individuals, involves amyloid depositing in the walls of larger leptomeningeal and cortical arteries. These two subtypes have different relationships to Alzheimer’s pathology: Type 1, capillary CAA, is more specifically linked to AD and to the APOE4 genotype, and it is associated with more active perivascular inflammation.
During life, most CAA is silent. Patients with CAA often have no symptoms, and the condition is discovered incidentally — either on a brain MRI or, posthumously, at autopsy. But when CAA becomes symptomatic, the consequences can be serious: transient episodes of confusion or focal neurological symptoms, recurrent headaches, seizures, and a pattern of cognitive decline. The symptomatic inflammatory form, called CAA-related inflammation (CAA-ri), is characterized by perivascular immune activation — the body’s immune cells attacking the amyloid-laden vessels — and this process disrupts the blood-brain barrier (BBB), the tightly regulated interface that normally keeps blood-borne molecules out of the brain.
Diagnosing CAA in living patients relies almost entirely on brain MRI. The updated Boston Criteria version 2.0 provides a framework for classifying probable CAA based on imaging findings: lobar microbleeds (tiny hemorrhages appearing as dark spots on susceptibility-weighted MRI sequences), cortical superficial siderosis (iron deposits along the brain surface from prior small hemorrhages), and severely enlarged perivascular spaces in specific brain regions. No blood test or spinal fluid marker can currently diagnose CAA with certainty in a living person, and definitive diagnosis requires brain tissue — which is why advances in imaging biomarkers are so important for this field.
ARIA: When Treatment Triggers the Problem
Enter ARIA. When anti-amyloid antibody therapies clear amyloid from plaques and from vessel walls, the mechanical and inflammatory stress on already-fragile CAA-affected vessels can cause visible abnormalities on MRI. Clinicians divide ARIA into two types based on what appears on imaging. ARIA-E refers to edema or effusion — swelling around blood vessels visible as bright signal on FLAIR MRI sequences. ARIA-H refers to hemorrhagic changes — microbleeds or superficial siderosis appearing as dark lesions on susceptibility-weighted imaging.
The majority of ARIA events detected on monitoring MRI scans are asymptomatic. The patient feels nothing, and the abnormalities resolve on their own, often within weeks to a few months. But a meaningful minority of patients develop symptomatic ARIA, which can include headache, confusion, dizziness, visual disturbances, focal neurological symptoms resembling a stroke, and in severe cases, seizures or hospitalization. Symptomatic ARIA requires drug hold and sometimes permanent discontinuation, which eliminates the therapeutic benefit the patient was just beginning to receive.
The rates of ARIA with the current generation of anti-amyloid antibodies are substantial. In the pivotal clinical trial of lecanemab, approximately 21 percent of treated patients developed ARIA-E and 36 percent developed ARIA-H at some point during treatment. With donanemab, similarly high rates were observed. These numbers are already high across the general trial population — but they are dramatically higher in specific subgroups.
The strongest risk factor for ARIA, by a considerable margin, is the APOE4 genotype. APOE4 is the most common genetic risk factor for late-onset Alzheimer’s disease, and carrying one copy raises the risk of developing Alzheimer’s roughly threefold. Carrying two copies — being a homozygote, designated APOE4/4 — raises the risk by eight to twelve times. In the anti-amyloid trials, APOE4/4 homozygotes face ARIA rates of 33 to 67 percent, depending on the drug and the dose. In the APOLLOE4 trial specifically designed to study this population, 32 percent of APOE4/4 participants with early Alzheimer’s already had at least one lobar microbleed at baseline — before receiving any treatment — reflecting how extensive the underlying vascular disease already is. After treatment, the vulnerability compounds dramatically.
Other risk factors for ARIA include higher antibody doses, the presence of pre-existing CAA on baseline MRI, and treatment earlier in the disease course. But APOE4 status dominates all of them. This genetic specificity strongly suggests there is a biology underlying ARIA vulnerability that is tied to APOE4’s effects on the vasculature — and understanding that biology might reveal how to mitigate the risk.
Why APOE4 Makes Blood Vessels So Vulnerable
The APOE4 protein — the product of the APOE4 gene — has several well-established functions in the brain, most notably in transporting cholesterol and facilitating the clearance of amyloid-beta from the brain and from blood vessel walls. APOE4 performs these functions less efficiently than the more common APOE3 variant, which is part of why APOE4 carriers accumulate more amyloid. But APOE4’s effects on blood vessels go beyond amyloid clearance.
APOE4 promotes BBB dysfunction through several mechanisms. It activates a cellular pathway involving matrix metalloproteinase-9 (MMP9), an enzyme that degrades the proteins holding the BBB’s tight junctions together. It is associated with the loss of pericytes — the specialized cells that wrap around blood vessel walls and are essential for BBB maintenance. APOE4 also shifts the endothelium toward a chronic low-grade inflammatory state, even before significant amyloid deposits have formed. The result, in APOE4 carriers, is a blood-brain barrier that is pre-existing compromised — more permeable, more inflamed, and more structurally fragile — before any drug or disease complication adds further stress.
This vascular vulnerability is not just theoretical. In human brain tissue, APOE4 carriers with high CAA burden show measurably reduced astrocyte end-foot coverage of blood vessels, impaired tight junctions, increased MMP9 activity, and lower PDGFRβ levels (a marker of pericyte health) in their cerebrospinal fluid. When anti-amyloid antibodies begin to strip amyloid from these already-vulnerable vessel walls, the resulting inflammatory response occurs on top of a compromised foundation.
The cPLA2 Hypothesis: A Proposed Molecular Culprit
This is where the new perspective paper enters with a specific mechanistic proposal. We hypothesize that an enzyme called cytosolic phospholipase A2 — cPLA2, encoded by the gene PLA2G4A — sits at the intersection of these vascular problems and may be a central driver of the vascular inflammation underlying CAA-ri and ARIA susceptibility in APOE4 carriers.
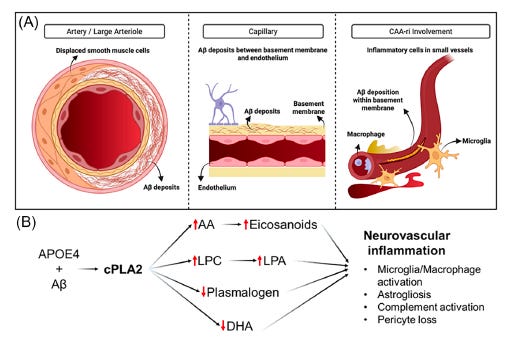
It is important to say upfront: this is a scientific hypothesis. The evidence marshaled in this perspective is compelling enough to justify serious investigation, but it does not yet prove that cPLA2 causes ARIA or that inhibiting it will prevent ARIA in humans. We argue that the existing evidence is strong enough to propose cPLA2 as a priority target and to outline how the hypothesis should be tested.
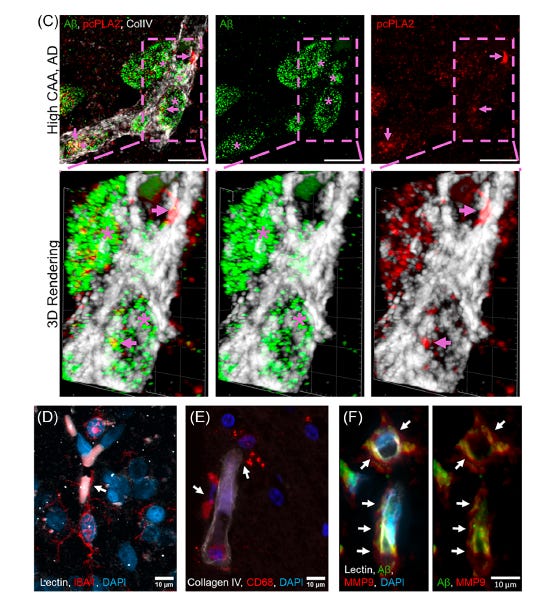
The figure above shows cPLA2 protein (red) covering the proximal areas to abeta (green) in blood vessels (white). Microglia and macrophages (brain immune cells) are lining up along the blood vessel, together with the enzyme MMP9, which breaks down the blood-brain barrier junctions. Credit to Bilal Kerman for creating these high-resolution confocal images.
So what is cPLA2, and why does it matter? cPLA2 is an enzyme that sits inside cells — including brain endothelial cells, astrocytes, microglia, and pericytes — and cleaves fat molecules from cell membranes. Specifically, it liberates arachidonic acid (AA) from membrane phospholipids when it is activated by calcium and inflammatory signals. AA is the starting material for a large family of pro-inflammatory signaling molecules called oxylipins, which include prostaglandins, leukotrienes, and hydroxyeicosatetraenoic acids (HETEs). These molecules recruit immune cells, activate MMP9 (which degrades the BBB), and amplify the inflammatory cascade.
But cPLA2’s effects are not purely through what it releases. There is also the matter of what it removes. The same membranes that contain arachidonic acid also contain protective lipids — including plasmalogens (a special type of fat with a vinyl-ether bond that confers antioxidant properties and structural integrity) and docosahexaenoic acid (DHA), the omega-3 fatty acid essential for neuronal membrane health. cPLA2, when overactive, depletes these protective lipids while simultaneously generating pro-inflammatory ones. It is, in effect, a molecular switch that simultaneously amplifies the fire and consumes the fireproofing.
Elevations in the downstream products of cPLA2 — particularly 12-HETE and 15-HETE, measurable in blood and cerebrospinal fluid — have been found in Alzheimer’s patients and correlate with microglial activation and cognitive decline. In APOE4 carriers and in APOE4 mouse models, cPLA2 activity and cPLA2 phosphorylation (the activated form of the enzyme) are measurably higher compared to non-APOE4 controls. These are associations, observed in tissue samples and animal models — they do not prove cPLA2 is the driver of disease, but they establish that the enzyme is more active in precisely the context where vascular vulnerability is greatest.
The perspective paper further points to data from human post-mortem brain tissue: in brains with high CAA burden (confirmed AD patients with CAA scores of 2 or 3 out of 3), activated cPLA2 clusters visibly in and around the blood vessel walls at the sites of amyloid deposition — seen in high-resolution three-dimensional microscopy images included in the paper. Additionally, the level of cPLA2 activity in the vessel walls correlates with evidence of BBB leakage measured by extravascular fibrinogen staining. APOE4 carriers with definite CAA show significantly higher perivascular cPLA2 activity than those without CAA, independent of amyloid and tau pathology.
Animal model experiments add mechanistic plausibility. Genetic knockout of PLA2G4A in mouse models of Alzheimer’s disease reduces neuroinflammation, improves learning and memory, and decreases premature death. Amyloid-oligomer-induced neurotoxicity in neurons can be partly blocked by cPLA2 inhibition in culture. In the E4FAD mouse model — a mouse carrying human APOE4 and familial AD mutations — pharmacological inhibition of cPLA2 reduced amyloid accumulation in vessel walls and reduced hemorrhagic load as measured by MRI. These experiments show that the pathway is tractable, but mice are imperfect models of human Alzheimer’s disease, and such findings do not guarantee that the same results will hold in human clinical trials.
The paper also situates cPLA2 within the complement cascade — the innate immune system’s rapid-response arm. In CAA, amyloid deposits in vessel walls activate complement through the classical pathway, and complement components including MAC (membrane attack complex) cause direct cell lysis. Components C5b-9 and C6, downstream of this cascade, are associated with subcortical hemorrhage and cortical superficial siderosis. Activation of complement by amyloid and APOE may further activate cPLA2, potentially creating a self-reinforcing cycle of vascular injury. Again, this remains mechanistically proposed — the convergence is biologically coherent but not yet causally proven in humans.
Why Previous Anti-Inflammatory Approaches Failed
One of the most instructive threads in the perspective paper is its analysis of why previous anti-inflammatory treatments have not worked in Alzheimer’s disease — and what that failure tells us about where to look next.
The most extensively studied approach has been COX inhibitors: drugs like aspirin, naproxen, and celecoxib, which block the cyclooxygenase enzymes that convert arachidonic acid into prostaglandins. The reasoning seemed sound: if prostaglandins cause inflammation, blocking their production should be protective. The Alzheimer’s Disease Anti-Inflammatory Prevention Trial (ADAPT) randomized over 2,500 cognitively normal older adults to naproxen, celecoxib, or placebo and followed them for two years — until the trial was halted early because both active treatment arms trended toward higher AD rates and showed worse performance on global cognitive scores compared to placebo.
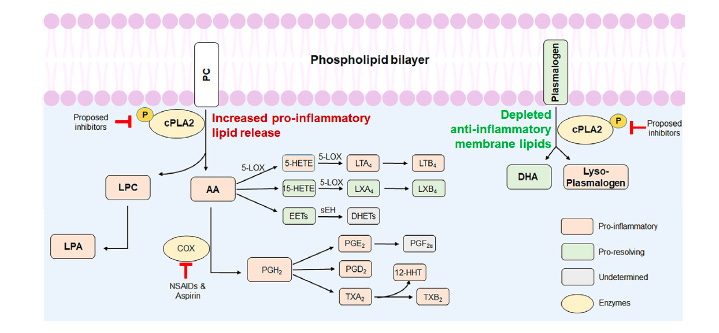
The failure of COX inhibitors is actually informative. These drugs work downstream of cPLA2 (As shown in Figure above), blocking only one branch of the arachidonic acid cascade. But when you block COX enzymes, the freed arachidonic acid is still being produced by cPLA2, and it can be redirected into the lipoxygenase (LOX) pathway, generating leukotrienes and HETEs — other pro-inflammatory oxylipins that COX inhibitors do not touch. You may also inhibit the COX-1 pathway, which produces some beneficial prostaglandins, and you leave intact the cytochrome P450 pathway, which generates yet another class of oxylipins. The result is a pharmacologically incomplete intervention that can cause compensatory activation of the very pathways it is trying to suppress. Furthermore, COX inhibitors have well-documented cardiovascular risks with long-term use, complicating their use in elderly patients.
The perspective paper argues that this history supports moving further upstream — to cPLA2 itself, which sits at the branch point before all these pathways diverge. Inhibiting cPLA2 would simultaneously suppress the lipoxygenase pathway, the COX pathway, and the production of lysophosphatidylcholine (LPC), while also — if the enzyme is sufficiently inhibited — preserving more of the protective membrane lipids that cPLA2 otherwise depletes. The logic is appealing. Whether it holds in human trials remains to be seen.
What Would It Take to Test This Hypothesis?
The perspective paper devotes considerable space to biomarker development and patient stratification — the infrastructure needed to actually test whether cPLA2 inhibition does anything useful in humans.
The proposed biomarker strategy has several layers. In the blood and cerebrospinal fluid, oxylipin profiling can measure the direct products of cPLA2 activity: PGE2, LTB4, 12-HETE, and 15-HETE. Elevated levels of these molecules in APOE4 carriers and their correlation with CAA severity and ARIA susceptibility would strengthen the hypothesis. Plasma oxylipin profiling offers a practical non-invasive readout; CSF sampling provides more direct access to what is happening near brain tissue.
Plasmalogen levels — the protective lipids depleted by overactive cPLA2 — offer a complementary readout. Plasmalogen deficiency is already recognized as a metabolic signature of Alzheimer’s disease progression. If cPLA2 inhibition restores membrane plasmalogens, this would provide mechanistic confirmation of target engagement in the right direction.
Imaging offers the most direct window into the brain. The paper describes PET tracers labeled with fluorine-18 attached to arachidonic acid and DHA. In preliminary studies in APOE4 knock-in mice, these tracers show elevated arachidonic acid uptake in regions consistent with heightened cPLA2 activity, particularly in cortical and perivascular regions. If these tracers can be validated in humans, they would allow clinicians to identify which patients have the most active cPLA2-mediated lipid dysregulation in their brain vessels, and to monitor directly whether a treatment is having an effect on that activity.
A clinical trial testing this hypothesis would ideally enrich for APOE4/4 homozygotes with imaging-confirmed CAA — precisely the patients at highest ARIA risk — and would measure oxylipin and plasmalogen biomarkers as primary pharmacodynamic endpoints before moving to clinical outcomes like ARIA incidence. The question at the center of such a trial would not be “does this drug slow cognitive decline?” (too hard, too long, too expensive for an early-phase trial) but rather “does inhibiting cPLA2 reduce the biochemical and imaging evidence of vascular inflammation in these patients?” That is a testable hypothesis with defined biomarkers and a realistic timeline.
Uncertainty and Future Directions
The cPLA2 hypothesis is scientifically well-constructed, biologically coherent, and supported by a convergence of associative and experimental evidence. It is not yet proven. The gap between a compelling mechanistic framework and a validated human therapeutic is wide, and the history of Alzheimer’s drug development is filled with hypotheses that survived every preclinical test and then failed in people.
Several specific uncertainties deserve emphasis. First, all the human tissue evidence is associative: elevated cPLA2 activity near amyloid deposits in vessel walls could reflect cPLA2 as a cause of vascular damage, or it could reflect cPLA2 activity as a consequence of damage caused by something else — or both, in a feedback loop. The causal arrow is not yet established.
Second, complete inhibition of cPLA2 could be problematic. cPLA2 has normal physiological roles in immune function, wound healing, and lipid metabolism. Loss-of-function mutations in humans cause a platelet dysfunction disorder and intestinal problems. The expectation is that partial inhibition — enough to dampen pathological vascular inflammation without eliminating normal immune signaling — will be both safe and effective, but this therapeutic window has not been established in humans.
Third, the oxylipin and plasmalogen biomarkers, while promising, have not yet been prospectively validated as predictors of ARIA risk in individual patients. The correlations observed in patient cohorts are encouraging, but whether a low-HETE or high-plasmalogen signature in a given patient’s blood or CSF reliably predicts that they will or won’t develop ARIA remains to be tested.
Looking ahead, the paper identifies three areas where progress is needed in parallel. Brain-penetrant cPLA2 inhibitors are the prerequisite — drugs that can reach the perivascular space in adequate concentrations, are selective for cPLA2 over related enzymes, and have a tolerable side effect profile with long-term use. (A companion paper in npj Drug Discovery describes early-stage compounds along these lines, which we covered in a previous post.) Biomarkers for target engagement must be validated — particularly plasma oxylipin profiling and the [18F]-labeled PET tracers — so that clinical trials can confirm drug activity before committing to large, expensive outcome studies. And the most immediate opportunity may lie in clinical integration with the existing anti-amyloid immunotherapy trials: APOE4/4 patients starting lecanemab or donanemab represent a precisely defined high-risk group where an ARIA-prevention trial could be conducted with a meaningful primary endpoint within 12 to 18 months.
The paper also raises an intriguing possibility about timing. Recent scholarship suggests there may be two distinct stages of inflammation in Alzheimer’s disease — an early, pre-clinical stage that is still amenable to anti-inflammatory intervention, and a later stage that has progressed beyond the reach of simple suppression. If that model is correct, earlier intervention targeting cPLA2 in presymptomatic APOE4 carriers with elevated amyloid burden might be far more effective than intervening after established CAA-ri.
Take-Home Messages
CAA — amyloid in the brain’s blood vessels — is nearly universal in Alzheimer’s disease, especially in APOE4 carriers. It is mostly silent, but it creates a vulnerable vascular environment that predisposes to ARIA when anti-amyloid therapies are given. APOE4/4 homozygotes face the highest burden of both CAA and ARIA, with rates of brain hemorrhage-related imaging abnormalities between 33% and 67% on current therapies.
Understanding why APOE4 carriers are so much more vulnerable requires understanding the specific molecular biology that makes their vessel walls fragile and inflamed. The new perspective paper proposes that the enzyme cPLA2 is a key upstream driver of that process, converting amyloid deposits in vessel walls into a chemical cascade of inflammatory lipid mediators while simultaneously depleting the protective fats that keep membranes resilient.
This is a hypothesis, not yet a proven mechanism. The evidence supporting it — from post-mortem human brain tissue, from mouse models, and from cell biology experiments — is substantial and convergent. But demonstrating causality in humans will require the development of validated biomarkers and a well-designed clinical trial in the right patient population.
The failure of COX inhibitors and aspirin in Alzheimer’s trials is reframed by this hypothesis not as evidence that inflammation doesn’t matter, but as evidence that targeting inflammation too far downstream is pharmacologically incomplete. Going upstream — to cPLA2 itself — offers a more comprehensive suppression of the pro-inflammatory oxylipin cascade, combined with the potential benefit of preserving protective membrane lipids.
The ultimate test of this idea will come from humans. We are working on candidate drugs that penetrate the brain in sufficient concentrations and inhibit cPLA2 with the potency and selectivity needed for a rigorous trial.
About the Paper
Kanwal A, Kerman BE, Wang S, Camey K, Li B, Flores-Aguilar L, Ali N, McIntire LB, Shu CA, Louie SG, Head E, Arvanitakis Z, Yassine HN. “A perspective: PLA2G4A as drug target for vascular inflammation in Alzheimer’s disease.” Alzheimer’s & Dementia. 2026;22:e71320. https://doi.org/10.1002/alz.71320
Ambreen Kanwal and Bilal E. Kerman are co-first authors.
Acknowledgments
This work was supported by the National Institute on Aging (RF1AG076124, R01AG055770, R01AG067063, R01AG054434, R21AG056518, P30AG066530, R01AG082362, P30AG10161, P30AG72975, and R01AG15819), the Alzheimer’s Drug Discovery Foundation (ADDF; GC-201711-2014197), donations from the Vranos and Tiny Foundations, and Ms. Lynne Nauss. Additional support was provided by the National Institutes of Health (P50AG05142, R01AG074549, R01AG078800, R01AG072794, and RF1AG059621).
Insightful. Thank you for this clear description of your hypothesis, research and the implications.
Wondering if plasmologens are good or bad for the brain