

The Amyloid Hypothesis of Alzheimer's Disease: What is Validated, What is Missed, and What Comes Next
One of the most debated hypotheses in modern medicine

The Birth of a Big Idea: Where the Amyloid Hypothesis Came From
To understand why the field of Alzheimer’s research has spent three decades pursuing a single protein, you need to appreciate how consistent the original clues were. In 1992, John Hardy and Gerald Higgins formally proposed what became known as the amyloid cascade hypothesis—the idea that the abnormal buildup of a protein fragment called amyloid-beta (Aβ) in the brain is the earliest and most critical trigger of Alzheimer’s disease (AD). This was not speculation. It was a framework built on converging lines of genetic evidence that, taken together, pointed toward amyloid as the initiating event.

The first and perhaps most powerful piece of evidence came from people with Down syndrome. Individuals with trisomy 21 carry three copies of chromosome 21, which also happens to be where the gene encoding the amyloid precursor protein (APP) resides. Because of this gene-dosage effect, people with Down syndrome produce more Aβ throughout their lives—and virtually all of them develop the hallmark plaques and tangles of Alzheimer’s pathology by their late thirties and forties. Those who die young from other causes in their early-to-mid teens already show diffuse amyloid deposits in the brain, with tangles and neurodegeneration following later. This natural experiment—triple the dose of APP, and you observe early, progressive Alzheimer’s pathology—provided a foundational biological anchor for the hypothesis.
The second line of evidence came from rare inherited forms of early-onset Alzheimer’s disease (familial AD, or FAD). Scientists discovered that mutations in the APP gene itself, or in proteins called presenilin-1 and presenilin-2 (the enzyme that cuts APP to generate Aβ), all caused aggressive Alzheimer’s in people in their forties and fifties. These mutations share a unifying biochemical consequence: they shift the balance of Aβ production toward longer, stickier, more aggregation-prone forms, particularly Aβ42. Strikingly, a rare protective mutation in the APP gene (A673T) was later identified in Icelandic families: carriers produced slightly less Aβ throughout their lives and were substantially protected from both Alzheimer’s dementia and age-related cognitive decline, with some showing very few amyloid plaques even at age 100. As Selkoe and Hardy wrote at the 25-year anniversary of the hypothesis, “definitive proof... could only come from clinical trials that selectively target Aβ and produce slowing and ultimately arrest of cognitive decline.” The hypothesis also gained important genetic credibility when the discovery of ApoE4 as a major risk factor for late-onset AD was found to act, at least in part, by impairing the brain’s ability to clear Aβ effectively. Taken together, these observations across Down syndrome, inherited mutations, protective variants, and population genetics built a coherent biological case: Aβ dyshomeostasis—too much production, too little clearance, or both—sits at the beginning of the Alzheimer’s disease cascade, preceding tangles, neurodegeneration, and cognitive decline by years or even decades.
Validation at Last: How Recent Clinical Trials Supported the Amyloid Cascade Hypothesis
The road from biological hypothesis to therapeutic validation was long and littered with failure. For roughly two decades, nearly every major pharmaceutical effort targeting amyloid came up empty. Anti-amyloid vaccines were halted due to dangerous brain inflammation. Drugs designed to block the enzymes that produce Aβ (beta- and gamma-secretase inhibitors) either failed to show benefit or actually worsened outcomes. Dozens of monoclonal antibodies—proteins designed to bind and clear Aβ from the brain—missed their primary endpoints in large Phase 3 trials, including failures for solanezumab, bapineuzumab, crenezumab, gantenerumab, and others. These repeated setbacks raised serious questions about whether targeting amyloid was a viable therapeutic strategy at all.
But the failures contained instructive lessons. They indicated that treating patients who already had moderate-to-severe dementia was likely too late, and that not all anti-amyloid antibodies were equivalent—those that powerfully cleared fibrillar plaque, the dense aggregated form most associated with downstream pathology, appeared more likely to produce measurable biological effects. Armed with these refinements, a new generation of trials targeting earlier disease stages with more potent plaque-clearing antibodies produced the long-awaited result. The CLARITY AD trial of lecanemab (2022) enrolled 1,795 patients with early symptomatic Alzheimer’s and demonstrated a statistically significant 27% slowing of clinical decline at 18 months, as measured by the Clinical Dementia Rating Sum of Boxes (CDR-SB). The TRAILBLAZER-ALZ 2 trial of donanemab (2023) enrolled 1,736 early-stage patients and showed a 35% slowing of decline. Both drugs not only cleared amyloid plaques robustly—they also produced reductions in phosphorylated tau (p-tau) biomarkers in blood and cerebrospinal fluid. This biomarker finding was among the most scientifically significant: if reducing Aβ also lowers downstream p-tau—a protein that becomes pathologically modified and spreads through the brain as a consequence of amyloid accumulation—it provides biological support for amyloid’s position upstream in the disease cascade, rather than as a parallel or unrelated process.
Not the Whole Story: Why Amyloid Doesn’t Fully Explain Alzheimer’s Dementia
Validating the amyloid cascade hypothesis is not the same as claiming it fully explains the clinical syndrome of Alzheimer’s dementia across all affected people. There are several important reasons to interpret the clinical trial results with measured expectations.
Even in the genetic forms of Alzheimer’s that originally anchored the hypothesis, the disease mechanism appears more complex than Aβ overproduction alone. In Down syndrome, the extra copy of APP on chromosome 21 was long assumed to cause AD primarily by increasing Aβ42 production. But work from Ralph Nixon’s laboratory has shown that the β-secretase-cleaved carboxyterminal fragment of APP (APP-βCTF, or C99)—generated from the same excess APP—directly elevates lysosomal pH by approximately 0.6 pH units in cells from individuals with Down syndrome, inactivating cathepsin D and other lysosomal hydrolases that depend on acidic conditions to function. This lysosomal dysfunction is detectable perinatally, preceding amyloid plaque formation by years, and is reversed by normalizing lysosomal pH or by reducing APP expression with siRNA or BACE1 inhibition. Similarly, in familial AD caused by PSEN1 mutations, the presenilin-1 protein has a role in lysosomal acidification entirely separate from its function in the γ-secretase complex that cleaves APP: PSEN1 acts as a chaperone for the v-ATPase V0a1 subunit and is required for proper lysosomal acidification. Disease-causing PSEN1 mutations impair this function, elevating lysosomal pH and creating a cascade of autophagic substrate accumulation and progressive neurodegeneration that Nixon and Rubinsztein characterize as a primary driver of AD pathogenesis, not merely a consequence of Aβ . These findings introduce a meaningful qualification: even in the most penetrant genetic forms of Alzheimer’s, the path from gene mutation to neurodegeneration may run through lysosomal failure in parallel with—or even upstream of—Aβ accumulation. The distinction between “amyloid-driven” and “endolysosomal-driven” pathology may be less clean than the original formulation assumed.
More than 99% of Alzheimer’s cases are nonetheless late-onset and sporadic, arising from the complex interaction of genetics, aging, lifestyle, vascular health, and environmental factors. In this much larger population, amyloid accumulation appears to be a contributing factor but not the sole sufficient cause of dementia, and other pathological processes operate in parallel.
The first consideration is the magnitude of clinical benefit from anti-amyloid therapies. The CDR-SB is a scale ranging from 0 (no impairment) to 18 (severe dementia), where clinicians rate how well a person manages six life domains: memory, orientation, judgment, community activities, home life, and personal care. In early Alzheimer’s, scores typically fall between 2 and 8. The minimum difference on this scale that most clinicians consider meaningful to a patient’s daily life is estimated at 1 to 2 points. Lecanemab slowed decline by approximately 0.5 points over 18 months relative to placebo; donanemab by approximately 0.7 points over roughly 20 months. In plain terms: treated patients declined somewhat more slowly, but both groups continued to decline, and the average difference between treated and untreated patients was below what most clinicians or caregivers would detect day to day. For any individual patient, the chance of experiencing a perceptible benefit is modest, and it must be weighed against the risk of ARIA—amyloid-related imaging abnormalities including brain swelling and microbleeds—that affected 13–37% of treated patients across the trials, with a subset requiring discontinuation or hospitalization. This does not diminish the scientific significance of a positive trial, which after decades of failure is genuinely important. It does, however, raise a practical and conceptual question: if amyloid is the central driver of Alzheimer’s dementia, why does substantially clearing it from the brain produce relatively small effects on the rate of cognitive decline? The most likely answer is that by the time patients are enrolled in these trials—even at the “early” stage—substantial neurodegeneration, tau pathology, and inflammation have already accumulated, and removing amyloid at that point is analogous to extinguishing a fire after the structure has burned. The disease has become self-sustaining through mechanisms that are no longer amyloid-dependent.
This complexity becomes further apparent when we examine racial and ethnic diversity in Alzheimer’s presentation. A 2026 study by Wheeler and colleagues examined 1,181 cognitively unimpaired and 383 mildly cognitively impaired participants from the Health and Aging Brain Study–Health Disparities (HABS-HD), encompassing non-Hispanic White (NHW), Hispanic, and Black older adults. Using tau PET imaging to map tau pathology in the medial temporal lobe (MTL)—a brain region critical for memory—and amyloid PET to characterize amyloid burden, the study revealed important group differences. After controlling for age, sex, and education, Black and Hispanic participants showed significantly higher MTL tau levels than NHW participants, even after accounting for choroid plexus off-target signal (a technical artifact that can artificially inflate tau measurements in certain brain regions). More notable still was the nature of the amyloid-tau-cognition relationship across groups: in NHW and Hispanic participants, amyloid positivity significantly amplified the relationship between tau and memory impairment—amyloid-positive individuals with high tau showed substantially lower cognitive scores. But in Black participants, this interaction between amyloid positivity and the tau-cognition relationship was attenuated and did not reach statistical significance (p = 0.067). Additionally, APOE4 significantly predicted higher MTL tau in NHW participants but showed no significant association with tau in Black or Hispanic participants. These findings suggest that in Black participants, factors other than amyloid and tau may be more dominant contributors to cognitive impairment—possibly cerebrovascular disease, greater burden of cardiometabolic risk factors like hypertension and diabetes, chronic stress exposure (which animal models show can promote tau hyperphosphorylation), or mixed brain pathologies. Prior neuropathological studies have shown that mixed pathology is more common in Black decedents with dementia. The amyloid cascade hypothesis, developed primarily from data in predominantly NHW populations, requires expansion to adequately account for the etiological diversity of Alzheimer’s dementia across different populations.
What Genetics Is Telling Us: Endolysosomal Dysfunction and Lipid Metabolism as Underlying Risk Factors
If amyloid does not fully account for Alzheimer’s dementia—particularly in late-onset, sporadic cases—where should we look for a more complete explanation? An important set of answers has been emerging from genome-wide association studies (GWAS), large-scale genetic analyses that identify common genetic variants associated with increased Alzheimer’s risk across tens of thousands of people. When researchers catalogued the genes most consistently implicated in late-onset AD risk, they found not a collection of genes involved primarily in amyloid production or clearance, but rather two distinct biological themes: lipid transport and metabolism, and endolysosomal trafficking—the cellular machinery responsible for sorting, recycling, and degrading molecular cargo inside cells.
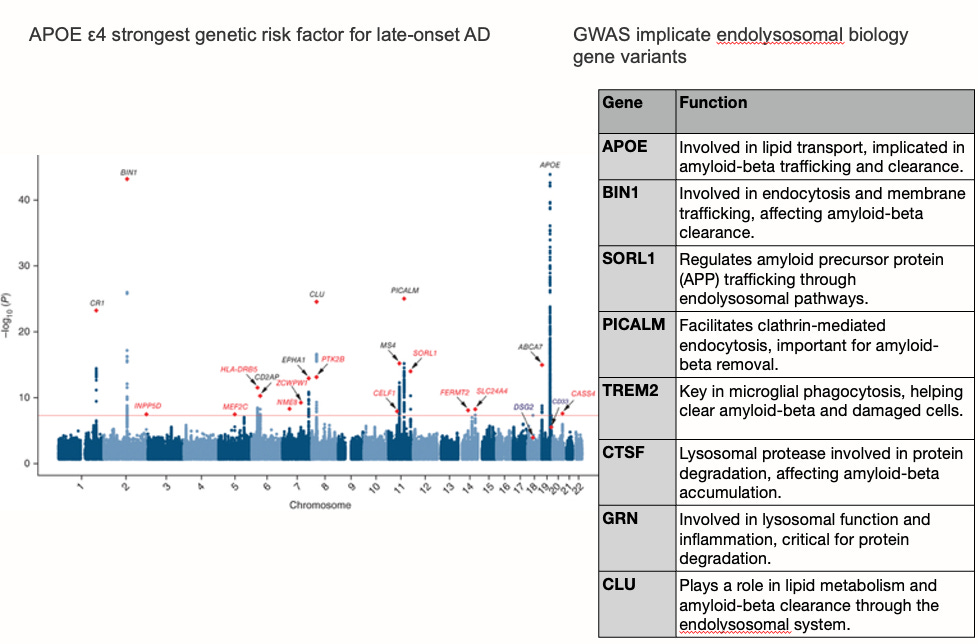
The landscape of late-onset GWAS genes reads like a chapter on cellular waste management and membrane biology. APOE itself—encoding the brain’s primary lipid carrier—represents by far the strongest genetic signal. SORL1 regulates the trafficking of amyloid precursor protein through endolysosomal compartments. BIN1 facilitates membrane trafficking and endocytosis. PICALM coordinates clathrin-mediated endocytosis relevant to Aβ removal. TREM2 is a receptor expressed by microglia—the brain’s immune cells—that governs their capacity to phagocytose cellular debris, including Aβ and damaged lipid-laden membranes. ABCA7, a lipid transporter closely related to ABCA1, carries loss-of-function mutations that substantially increase AD risk, likely by impairing cellular lipid efflux. CLU (clusterin) participates in lipid metabolism and Aβ clearance. GRN (granulin) is required for lysosomal function and inflammatory regulation. CTSF is a lysosomal protease involved in intracellular protein degradation. Taken together, these genes point consistently toward a shared biological pathway: the endolysosomal system—the cell’s internal sorting and disposal apparatus—is disturbed in Alzheimer’s disease in ways that may operate in parallel to, or upstream of, amyloid accumulation. The enlargement of early endosomes is among the earliest cytopathological features of AD, observable years before plaques appear, and is exacerbated by APOE4. This pattern suggests a complementary framework: that failure of endolysosomal recycling (Figure below), particularly in the astrocytes and microglia responsible for lipid transport and waste clearance, represents a biologically distinct pathway to Alzheimer’s pathology—one that the amyloid hypothesis was not designed to capture. Here, we can conclude that amyloid contributes to the endolysosomal recycling failure but is one of many drivers, and vice versa, endolysosomal failure accelerates amyloid accumulation.
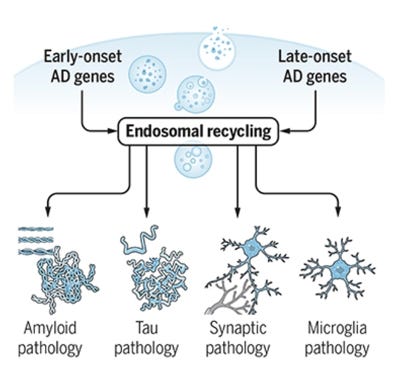
Figure from Scott and Petsko, Science Translational Medicine, 2020
APOE4: A Gene With Many Faces, Neuroinflammation Foremost Among Them
Of all the genetic factors in late-onset Alzheimer’s disease, none has a larger population-level impact than APOE4. Carrying one copy of the APOE4 allele roughly triples the lifetime risk of Alzheimer’s; carrying two copies increases risk more than tenfold. The initial explanation for this risk centered on amyloid: APOE4 impairs the brain’s ability to clear Aβ, contributing to earlier and more abundant plaque accumulation. That relationship is well-supported. But APOE4’s contribution to Alzheimer’s risk involves multiple interconnected biological mechanisms—organized around three distinct but overlapping “hits” on brain health.
The first hit is disrupted lipid handling. ApoE is the brain’s primary lipid transport protein, produced mainly by astrocytes and microglia to deliver cholesterol and other lipids essential for synaptic maintenance and membrane repair. The APOE4 isoform has an altered protein conformation that reduces its lipid-loading efficiency, particularly through a transporter called ABCA1. Research from our lab demonstrated that in APOE4 astrocytes, ABCA1 becomes abnormally retained in lysosomes rather than recycling to the cell surface, impairing the formation of small, lipidated HDL particles that are associated with protection against amyloid accumulation. This lysosomal sequestration is driven in part by co-aggregation of ApoE4 and ABCA1 proteins in an environment enriched with oxysterols (oxidized cholesterol metabolites), which are elevated in APOE4 brain tissue. The resulting cholesterol dyshomeostasis sets off a compensatory over-expression of ABCA1—which then activates mTORC1 in lysosomes, triggering cellular senescence in astrocytes and microglia. Senescent brain cells surrounding amyloid plaques have been observed in human AD brain tissue, staining positive for lipofuscin and GPNMB (classical senescence markers), suggesting that APOE4-driven lipid dysregulation accelerates cellular aging in the very cells whose function is to protect neurons.
The second hit is neurovascular inflammation. APOE4-expressing microglia are oriented toward a chronically activated, pro-inflammatory state. In the brain, APOE4 amplifies activation of the classical complement cascade—a molecular immune recognition system—which, when chronically overactivated, accelerates excessive synaptic pruning by microglia and promotes neurodegeneration. APOE4 also upregulates matrix metalloproteinase-9 (MMP9), an enzyme that degrades the tight junctions of the blood-brain barrier, allowing inflammatory molecules to enter the brain parenchyma and fibrinogen to accumulate near neurons. These neurovascular effects appear to operate at least partly independently of amyloid pathology and may help explain why Alzheimer’s dementia in APOE4 carriers has been more difficult to treat with amyloid-targeted therapies alone.
The third hit is neuronal dysfunction, including impaired insulin receptor trafficking (APOE4 sequesters insulin receptors in early endosomes), reduced apoE receptor signaling important for synaptic plasticity, and mitochondrial vulnerability. Together, these three hits illustrate why clearing amyloid from an APOE4 carrier’s brain may not be sufficient to reverse or halt dementia: the biological disruption involves lipid pathways, immune activation, and vascular integrity in ways that extend well beyond what the amyloid hypothesis alone was designed to address.
What We Still Don’t Know—and Where the Science Is Heading
Three decades after the amyloid hypothesis was formalized, the field of Alzheimer’s research is confronting a more complex biological reality than the original framework anticipated. Evidence now supports that amyloid is an important upstream contributor to Alzheimer’s pathology—at least in the populations and disease stages studied to date. But several major uncertainties limit our ability to translate this understanding into broadly effective therapies.
A central open question concerns treating people before symptoms develop. The trajectory of Alzheimer’s pathology is now understood to unfold over 15 to 20 years before cognitive symptoms appear, with amyloid accumulating silently in the brain throughout midlife and late life. This raises a logical but practically challenging question: would removing amyloid at this pre-symptomatic stage—before tau pathology spreads and neurons begin to die—produce substantially larger clinical benefits? The A4 (Anti-Amyloid Treatment in Asymptomatic Alzheimer’s) trial tested this directly, enrolling cognitively normal older adults with elevated amyloid on PET imaging and treating them with solanezumab for 4.5 years. The result was negative—no significant slowing of cognitive decline—though solanezumab was a relatively weak amyloid-clearing agent. Ongoing trials are now testing more potent antibodies, including lecanemab, in cognitively normal individuals with elevated amyloid (the AHEAD 3-45 and A45 trials). The scientific rationale is sound, but the practical challenges are formidable. Because the base rate of progression from amyloid positivity to dementia is low in any given multi-year window, extremely large trials over many years are required to detect a meaningful difference—and most participants who receive treatment (and its risks) may not have developed dementia within the trial period regardless. Identifying who carries silent amyloid accumulation at scale requires either amyloid PET imaging, which is expensive and not universally accessible, or emerging blood-based biomarkers for Aβ and phospho-tau that are still being validated for broad clinical use. The ARIA risks from potent anti-amyloid antibodies in a population that currently has no symptoms—and no guarantee they would develop dementia—raises difficult questions about acceptable risk-benefit balance. Prevention is the intellectually compelling endpoint for the amyloid hypothesis, but achieving it equitably and safely remains one of the field’s most pressing unresolved challenges.
Beyond prevention, significant uncertainty remains about which forms of Aβ and tau are most toxic, and through what precise molecular mechanisms. We do not yet adequately understand how amyloid pathology interacts with cerebrovascular disease, neuroinflammation, and metabolic disruption in the complex mix of causes that characterizes most late-onset dementia. We do not know why amyloid appears to drive tau pathology with different efficiency across racial and ethnic populations, or whether ancestral differences in APOE4 haplotype structure (documented across African and Latin American populations) modify the downstream consequences of amyloid accumulation. We do not yet know whether therapies targeting the endolysosomal system, APOE4 lipidation, neuroinflammation, or cholesterol metabolism will demonstrate clinical efficacy—though first-generation therapies directly targeting APOE4 biology, including APOE4 structure correctors that convert APOE4 toward a less pathogenic conformation and lipidation-enhancing agents designed to restore ABCA1-mediated cholesterol transport, are now in early clinical testing. And we face a persistent practical dilemma: the therapies currently approved for disease modification are expensive, require intravenous infusion, carry meaningful safety risks, and provide modest average benefit that may not be detectable in individual patients.
Take-Home Messages
The amyloid cascade hypothesis has now been clinically validated. Anti-amyloid clinical trials demonstrated that clearing Aβ from the brain slows cognitive decline and simultaneously lowers downstream phospho-tau biomarkers—supporting the position of amyloid as an upstream driver in the disease cascade.
The clinical benefits of current anti-amyloid therapies are real but modest. Lecanemab and donanemab slowed decline by approximately 0.5–0.7 points on an 18-point scale over 18–20 months—below the threshold many clinicians consider perceptible to patients and caregivers. Both groups continued to decline. This likely reflects that by the time patients enroll in trials, the disease has progressed beyond amyloid dependence.
Average trial results do not tell the whole story: some individuals may respond meaningfully. Population averages can obscure the fact that a subset of patients may derive substantially greater benefit from anti-amyloid therapy—particularly those treated earliest, with lower tau burden. Identifying who is most likely to respond is an active and critical area of research. These approvals represent the first disease-modifying treatments in Alzheimer's history and reflect decades of hard-won scientific progress that should not be minimized.
Amyloid may be necessary but not sufficient for dementia. In late-onset, sporadic Alzheimer’s—which represents over 99% of cases—amyloid accumulation interacts with vascular disease, mixed pathology, neuroinflammation, and other factors that the amyloid hypothesis alone does not fully account for.
Alzheimer’s dementia is not the same disease in all populations. Black participants showed a distinct relationship between amyloid, tau, and cognition compared to non-Hispanic White participants, suggesting that other biological or sociostructural factors may be primary contributors to cognitive decline in this group. The vast majority of foundational Alzheimer’s research has been conducted in predominantly White cohorts.
Late-onset GWAS genes tell a story that extends beyond amyloid. Risk variants in APOE, BIN1, SORL1, PICALM, TREM2, and ABCA7 collectively implicate endolysosomal dysfunction and lipid metabolism as major disease pathways—suggesting that cellular waste clearance and lipid transport are as central to AD risk as amyloid dynamics, and open the path for new drug targets.
APOE4 drives Alzheimer’s risk through at least three parallel mechanisms: disrupted lipid handling and HDL formation, neurovascular inflammation and blood-brain barrier disruption, and direct neuronal dysfunction. Anti-amyloid therapies may be less effective in APOE4 carriers partly because they do not address these additional pathways.
Neuroinflammation is a central and potentially modifiable driver of disease, particularly in APOE4 carriers. Chronically activated microglia, primed by lipid dysregulation, may propagate tau pathology and neurodegeneration independently of ongoing amyloid accumulation.
Prevention is the most scientifically compelling application of the amyloid hypothesis, but also the most difficult to achieve. Treating cognitively normal individuals with elevated amyloid requires very large, long trials; effective and accessible screening tools; and a careful reassessment of acceptable risk-benefit balance in a population that currently has no symptoms.
The next generation of Alzheimer’s therapies will likely need to be combinatorial—addressing amyloid and tau alongside lipid metabolism, endolysosomal function, neuroinflammation, and cerebrovascular health. The validation of the amyloid hypothesis is a scientific milestone, but the path to broadly effective dementia prevention will require a substantially broader biological framework.
This is a thorough and well-written review of the evidence that supports this hypothesis. However, I think that you oversimplified a bit and missed the big problem, which is disease heterogeneity. You correctly state the obvious, that some patients respond much better than the average response, but neglect to mention that a significant number of patients do not respond at all (why such a big difference?). The trials of the existing drugs had to be huge (900 per arm!) and extremely expensive in order to average out the variability and arrive at a statistically significant result. This is not a sustainable model for development of new AD drugs. The problem is that Alzheimer's disease is not a single entity. We need to tackle this issue before we discuss novel targets or combination therapies.
Also, maybe you should publish this in a "traditional" journal too after elaborating on some more details.
Will be glad to review it 😇