

cPLA2 and the Synapse
New human data on an inflammatory enzyme linked to cognitive impairment in Alzheimer's disease

In earlier work, we examined why omega-3 supplementation failed to produce consistent cognitive benefits in Alzheimer’s disease, particularly among APOE4 carriers. That work shifted our focus from simple lipid deficiency toward altered lipid dynamics within brain cells. Across blood, cerebrospinal fluid, and brain tissue, we observed dysregulated omega-3–to–omega-6 balance and increased arachidonic acid–derived inflammatory signaling.
Repeatedly, one enzyme emerged as central to that biology: calcium-dependent phospholipase A2 (cPLA2).
The remaining question was both anatomical and clinical:
Is cPLA2 activated at human synapses—the structures most closely linked to cognition—and is that activation associated with impairment?
Our recent study was designed to address that question directly. Dr. Qiulan Ma led this study at USC over the past 3 years.
What Is a Synaptosome?
Synapses are the specialized junctions where neurons communicate. Synaptic density and integrity are among the strongest pathological correlates of cognitive function in Alzheimer’s disease.
Because living human synapses cannot be directly studied, we isolate synaptosomes from postmortem brain tissue. Synaptosomes are subcellular particles enriched in intact presynaptic and postsynaptic components. They preserve synaptic membranes, receptors, scaffold proteins, and signaling enzymes.
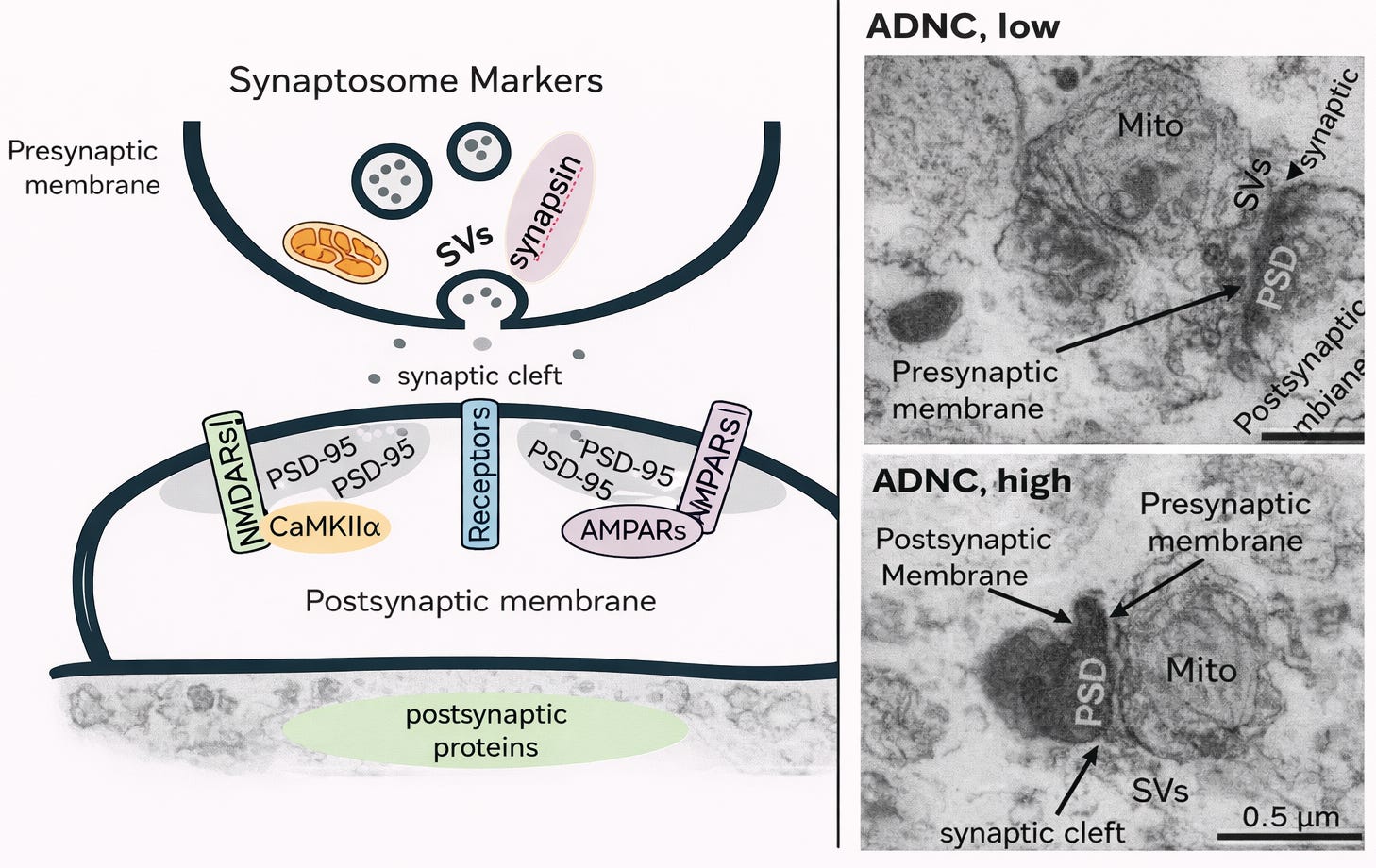
Figure 1. Synaptosome markers and corresponding human synaptic ultrastructure.
Left: Simplified schematic of a human synaptosome illustrating preserved presynaptic and postsynaptic components. The presynaptic membrane (upper thick dark line) contains SVs (synaptic vesicles) and synaptic proteins including synapsin, as well as mitochondria that support neurotransmission. The synaptic cleft is the extracellular space between the pre- and postsynaptic membranes. The postsynaptic membrane (lower thick dark line) contains key excitatory synaptic markers including NMDARs (N-methyl-D-aspartate receptors), AMPARs (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors), PSD-95 (postsynaptic density protein 95), and CaMKIIα (calcium/calmodulin-dependent protein kinase II alpha), which are central to synaptic signaling and plasticity.
Right: Representative transmission electron microscopy images of isolated human synapses from brains with low and high Alzheimer’s disease neuropathologic change (ADNC). Labeled structures include Mito (mitochondria), SVs (synaptic vesicles), PSD (postsynaptic density), presynaptic membrane, postsynaptic membrane, and the synaptic cleft. Scale bar = 0.5 μm.Although not complete neurons, they retain the biochemical machinery of synapses and allow direct measurement of synaptic protein levels and lipid mediators.
Using synaptosomes from participants in a longitudinal aging cohort with detailed cognitive assessments prior to death (Religous Order Study), we examined cPLA2 levels, downstream lipid signaling, and their relationship to cognition.
Selective Synaptic Vulnerability
Synaptosome yield was significantly reduced in individuals with mild cognitive impairment (MCI) and Alzheimer’s dementia compared with cognitively normal controls. In contrast, glial- and myelin-enriched fractions were not similarly reduced.
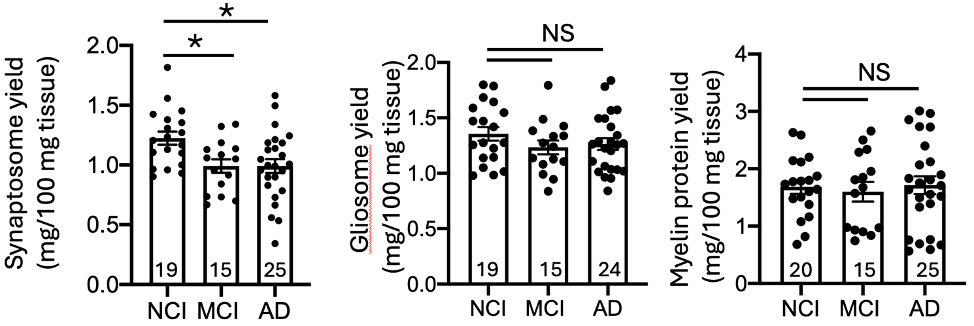
Fig. 2 Reduction of synaptosome yield in MCI and AD cases.
This finding supports selective synaptic vulnerability during disease progression and confirms that synaptic loss is detectable even at the MCI stage.
Elevated cPLA2 at Human Synapses
We measured two isoforms: cPLA2α and cPLA2β.
Both were elevated in synaptosomes from Alzheimer’s dementia cases. Notably, cPLA2β was already elevated in MCI.
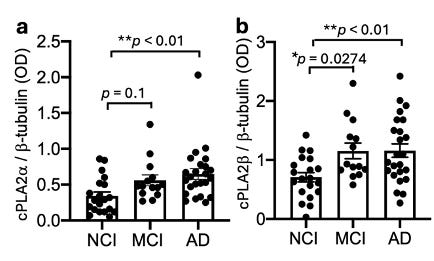
Fig. 3 Elevation of cPLA2 in synaptosomes of AD.
This suggests that synaptic cPLA2 activation is not restricted to advanced disease and may emerge during earlier clinical phases.
Association With Cognitive Performance
Higher synaptosomal cPLA2β levels were associated with worse global cognition and episodic memory after adjusting for age, sex, and education. These associations were particularly strong in male participants.
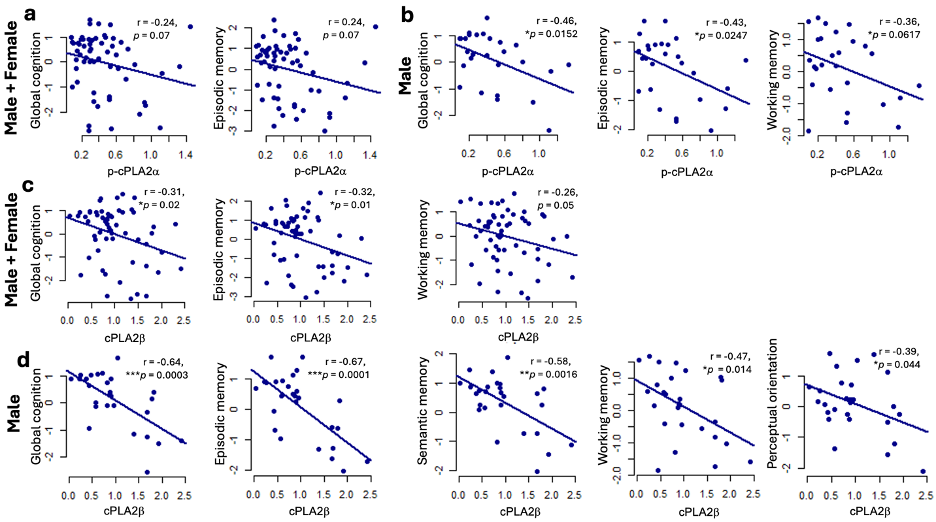
Fig. 4 Correlation between synaptosomal cPLA2 levels and cognitive dysfunction. a. p-cPLA2a (all). b. p-cPLA2a (male participants). c. cPLA2b (all). d. cPLA2b (male participants). Residuals were calculated from linear regression models in which cognitive function was regressed on age at last visit, sex and education. For sex-based analyses, residuals were calculated from models regressing cognitive function on age at last visit and education. *P < 0.05, **P < 0.01. ****P < 0.0001. ROS Cohort.
These findings are correlational and do not establish causation. However, they demonstrate that synaptic cPLA2 levels closely align with clinical impairment in humans.
Biochemical Evidence of Pathway Activation
cPLA2 hydrolyzes membrane phospholipids to release arachidonic acid (AA), which is then converted into eicosanoids—prostaglandins that regulate inflammatory signaling.
In Alzheimer’s synaptosomes, we observed:
Increased arachidonic acid
Increased AA-derived eicosanoids
Strong correlations between cPLA2 levels and AA metabolites
Other lipid pathways, including cholesterol metabolism, were not similarly altered. This supports the specificity of the cPLA2–arachidonic acid pathway.
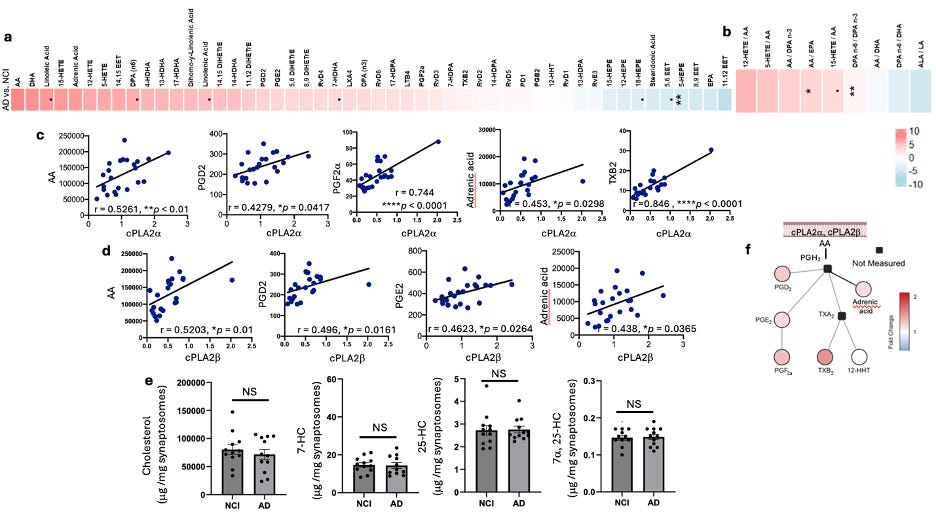
Fig 5. Alterations of AA and AA metabolites but not cholesterol and oxysterols in AD synaptosomes. a, b. Heatmap generated from lipidomic analysis of PUFAs and PUFAs’ metabolites in synaptosomes comparing AD vs. NCI by Wilcox rank sum test. The color represents the Wilcox test estimate, where redness indicated the increased lipid species in AD, and vice versa. Overall, relative to NCI, AD showed high arachidonic acid (AA) and AA metabolites, including increased TXB2 and PGF2a, which were highly corelated with cPLA2a (c). AA, PGD2, PGE2, and adrenic acid were corelated with both cPLA2a (c) and cPLA2b (d). Cholesterol and oxysterols, including 7-dehydrocholesterol (7-HC), 25-hydroxycholesterol (25-HC), and 7a, 25-hydroxychlesterol (7a, 25-HC), were not changed (e). f. A diagram of cPLA2 activation involved AA metabolic pathway. c-d Graphs show Pearson correlation between two variables, as described on the axis, *P < 0.05, **P < 0.01, ****P < 0.0001. ROS cohort.
These findings indicate that synaptic cPLA2 is not only elevated but enzymatically active.
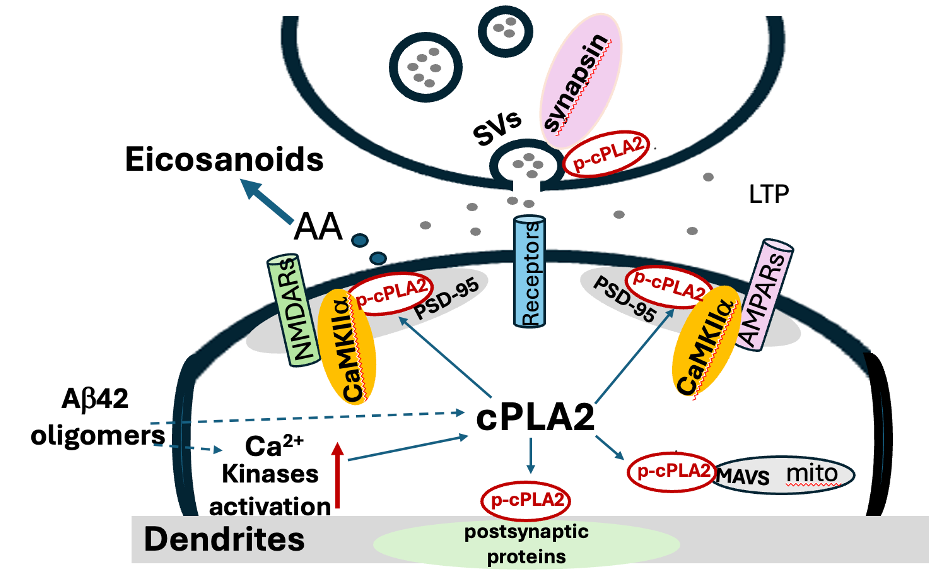
Excitotoxicity and Postsynaptic Localization
Excitotoxicity refers to neuronal injury resulting from sustained activation of excitatory glutamate receptors and excessive calcium influx. Elevated intracellular calcium activates downstream enzymes that destabilize synaptic structure.
cPLA2 is calcium-dependent.
In human iPSC-derived neurons, amyloid-β oligomers increased phosphorylation of cPLA2 and induced its redistribution to dendritic membranes. Activated cPLA2 colocalized with PSD-95 and CaMKIIα—proteins central to postsynaptic organization and synaptic plasticity.
Inhibition of cPLA2 suppressed amyloid-induced synaptic alterations in these models.
These observations support a mechanistic framework in which pathological calcium signaling activates cPLA2 at postsynaptic sites, leading to localized inflammatory lipid release and synaptic destabilization.
Interpretation and Uncertainty
Several limitations are essential to acknowledge:
Correlation does not establish causation.
Elevated synaptic cPLA2 may contribute to dysfunction, but it may also represent a downstream response to amyloid, tau, or other pathological processes.
Cross-sectional human data cannot determine temporal sequence.
We cannot establish whether synaptic cPLA2 activation precedes cognitive decline or follows it.
Experimental models simplify disease biology.
Cell-based systems allow mechanistic interrogation but do not replicate the full complexity of aging human brain networks.
Physiological roles of cPLA2 must be preserved.
cPLA2 participates in normal membrane remodeling and signaling. Excessive inhibition could disrupt essential processes.
These uncertainties require that translational claims remain cautious.
Why These Findings Support Testing a Disease-Modifying Hypothesis
A disease-modifying therapy alters the biological trajectory of a disease rather than temporarily improving symptoms.
The present study demonstrates that:
cPLA2 activation occurs directly at human synapses
Synaptic cPLA2 levels are associated with cognitive impairment
The arachidonic acid inflammatory pathway is upregulated at those sites
Amyloid-induced cPLA2 activation and synaptic alterations can be pharmacologically suppressed in human neurons
In our prior inhibitor study, selective brain-penetrant cPLA2 inhibition reduced amyloid-induced tau hyperphosphorylation and preserved synaptic markers in human neuronal systems. Those findings established that this pathway is modifiable and that its modulation affects downstream tau and synaptic biology.
Taken together, the data support a specific mechanistic proposition:
Pathological activation of cPLA2 may represent a convergence point linking lipid dysregulation, excitotoxic signaling, tau phosphorylation, and synaptic loss.
If correct, then selective modulation of cPLA2 activity—particularly during early clinical stages—could alter the rate of synaptic deterioration. That possibility meets the conceptual definition of a disease-modifying strategy.
This hypothesis has not yet been tested in clinical trials. It requires rigorous evaluation of safety, timing, and long-term impact.
However, the convergence of human synaptic data and pharmacologic modulation provides a biologically coherent framework for such testing.
Conclusion
This study establishes that cPLA2 activation occurs at human synapses in Alzheimer’s disease and is associated with cognitive impairment.
It connects earlier observations of lipid imbalance to a defined synaptic mechanism and links that mechanism to tau and excitatory signaling pathways known to drive neurodegeneration.
These findings do not establish therapeutic efficacy. They provide mechanistic support for evaluating whether selective, brain-penetrant modulation of cPLA2 can slow synaptic decline and modify disease progression.
The next step is careful translational testing.
Reference:
Ma, QL., Ebright, B., Li, B. et al. Evidence for cPLA2 activation in Alzheimer’s disease synaptic pathology. acta neuropathol commun (2026). https://doi.org/10.1186/s40478-025-02214-6
Funding:
This work was support by grants from by the National Institute on Aging. R01AG076124 (HNY, ZA), P30AG066530 to HNY; P30AG066530 subaward (QLM); R21AG089611(QLM) P30AG066530 to Neuropathology Core, R01AG070255 (AL), ROS is supported by P30AG10161, P30AG72975, and R01AG15819, and U01AG094622 to HNY and SL.
ROS resources can be requested at
https://www.radc.rush.edu
and www.synpase.org.
This is a high-signal post because it connects a “lipid enzyme” to the unit that actually predicts cognition: synaptic integrity.
cPLA2 (PLA2G4A) sits at a potent junction: it liberates arachidonic acid from membrane phospholipids, feeding downstream eicosanoid signaling and inflammatory amplification. In the brain, that’s not an abstract pathway; AA mobilization can directly alter membrane properties, receptor trafficking, and excitatory balance at synapses, which is exactly where Alzheimer’s phenotypes emerge earliest.
What makes this especially compelling is that the story is becoming increasingly human-anchored, not just mechanistic:
1. Recent human tissue work links higher cPLA2 activation/levels in AD brains to synaptic loss/neuritic plaque regions and neurodegeneration, consistent with cPLA2 as a contributor to cognitive decline rather than a bystander. 
2. Independent reports suggest altered cPLA2 isoforms in synaptosome-enriched fractions in MCI/AD, again pointing to synaptic-localized biology. 
3. And therapeutically, the field is moving from “interesting enzyme” to “druggable target,” with newer selective inhibitor development aimed at neuroinflammation in AD and related disorders. 
The nuance I appreciate is that this frames inflammation not as a vague cloud, but as membrane vulnerability + signaling bias at the synapse. It also makes the APOE4 angle feel less like fate and more like a mechanistic vulnerability worth targeting (lipid handling → synaptic stress → inflammatory amplification).
If anything, this pathway feels like a strong candidate for the next wave of “precision neuroinflammation”: not blanket suppression, but context-specific modulation of lipid signaling where synapses are failing first.
Really thoughtful synthesis!
This has opened my eyes to my own field of psychiatry...certain drugs increase cPLA2 activity, and others decrease it. Could we be prescribing drugs to patients who, unknown to us, are APOE4 carriers, and thus be "tipping them" even further into the flames of cPLA2 overdrive?