

ApoE directed Therapeutics in Alzheimer's disease, SFN 2025

Alzheimer’s disease has historically been viewed through the lens of amyloid plaques and tau tangles, but accumulating evidence shows that these features alone cannot fully explain why some individuals develop symptoms earlier or decline more rapidly. In the article, we highlight the powerful influence of the APOE gene—particularly the APOE4 variant—as a major upstream driver of Alzheimer’s vulnerability. APOE normally regulates lipid movement within the brain, modulates inflammation, and maintains the integrity of the blood–brain barrier. APOE4 disrupts these functions in ways that reshape the brain’s biochemical environment long before classical pathology becomes pronounced. The framework presented in the paper conceptualizes these disruptions as a series of three interconnected “hits”: early disturbances in lipid handling, followed by persistent neurovascular inflammation, and ultimately progressive neuronal dysfunction.
The first hit involves APOE4-induced changes in lipid metabolism within astrocytes and microglia. Studies, including those described in this work, show that APOE4 alters intracellular cholesterol trafficking, causing lipid accumulation in lysosomes and lipid droplets. These abnormalities activate stress pathways, impair cellular recycling mechanisms, and heighten inflammatory signaling. As metabolic pressure increases, glial cells—normally responsible for repair and maintenance—become less efficient at clearing debris and more prone to entering reactive states. This early lipid imbalance is increasingly recognized not merely as a downstream consequence of Alzheimer’s pathology, but as a primary biochemical shift that increases the brain’s susceptibility to later degeneration.
As these changes persist, the second hit emerges: chronic inflammation combined with vascular dysfunction. In APOE4 carriers, microglia adopt a more persistent proinflammatory profile, producing cytokines and lipid mediators that amplify neuronal stress. Complement activation, cPLA2 signaling, and other inflammatory pathways become excessively engaged. At the vascular level, APOE4 compromises the blood–brain barrier, allowing blood-derived proteins such as fibrinogen to leak into the brain and trigger additional glial activation. This combination of glial reactivity and vascular breakdown creates a self-reinforcing cycle that accelerates amyloid and tau propagation and destabilizes synapses.
These metabolic and inflammatory stresses ultimately converge on neurons, forming the third hit. Neurons in APOE4 carriers face reduced lipid availability, impaired synaptic receptor trafficking, altered energy homeostasis, and chronic exposure to inflammatory mediators. Receptors essential for synaptic plasticity may become trapped inside the cell, and deficits in autophagic clearance exacerbate cellular strain. Even in the absence of high amyloid or tau burden, these processes can erode neuronal connectivity and resilience. This underscores a broader point: APOE4 acts not just as a modifier of amyloid pathways but as an independent driver of neuronal vulnerability.
The therapeutic section of the article builds on this mechanistic understanding. A range of emerging APOE-focused interventions is highlighted, including structure correctors that stabilize APOE4’s conformation, compounds that enhance APOE lipidation, inhibitors of inflammatory pathways such as cPLA2 or complement, and therapies designed to strengthen the blood–brain barrier. More advanced approaches—such as antisense oligonucleotides that selectively reduce APOE4 expression, gene therapies introducing protective variants like APOE2, and cell type–specific epigenetic modulation—offer potential ways to intervene earlier and more precisely in the disease process. These strategies complement, rather than replace, amyloid- and tau-directed treatments by targeting the upstream biological disturbances most prominent in APOE4 carriers.
The conceptual figure 1 (credit to Cristelle Hugo) included in the article visually integrates these ideas.
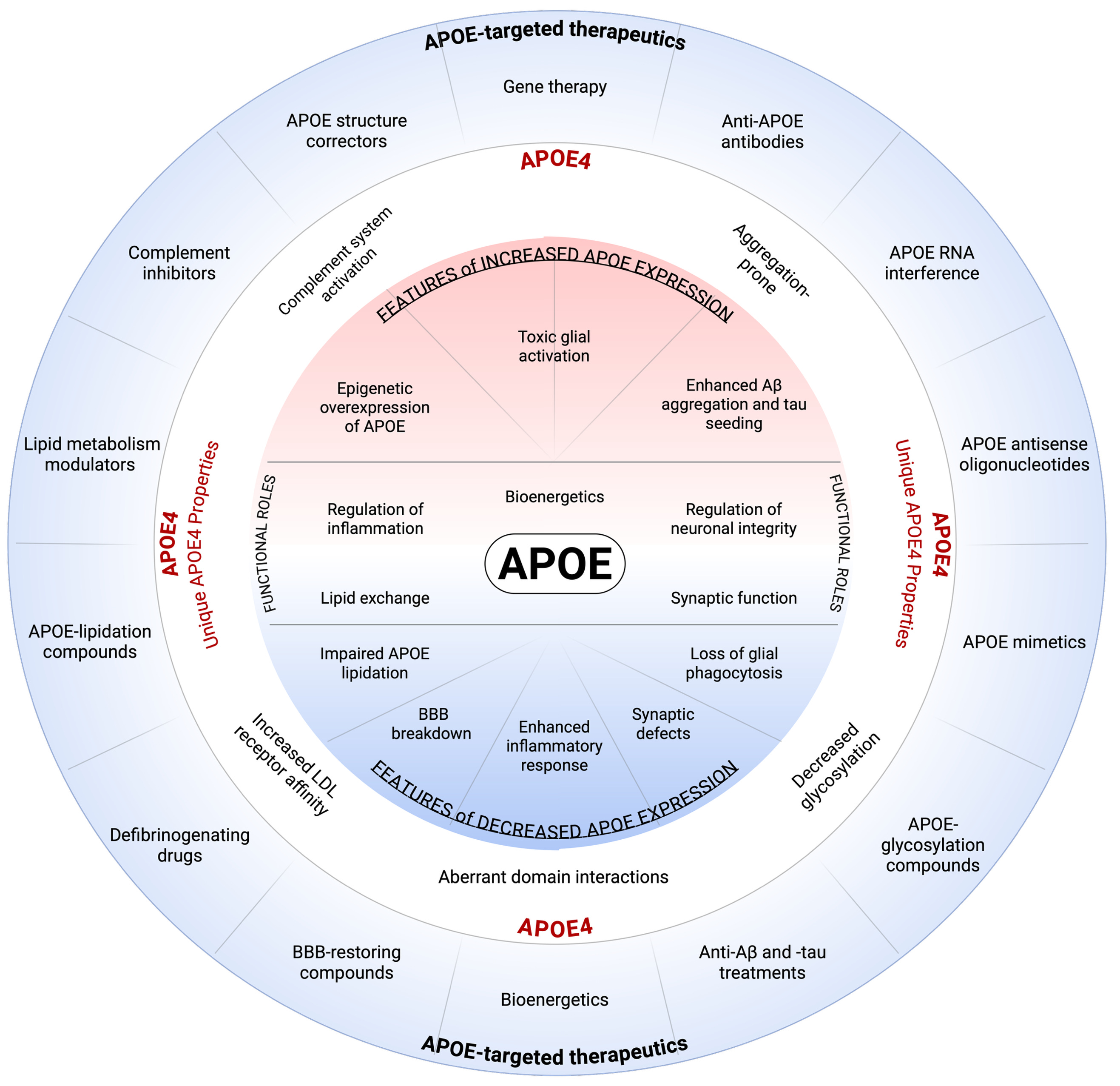
At the center are APOE’s essential physiological functions—lipid exchange, inflammatory regulation, synaptic maintenance, bioenergetics, and neuronal integrity. Surrounding this core, the diagram depicts how APOE4 disrupts these processes through both excessive and insufficient APOE activity: toxic glial activation, impaired lipidation, complement overactivation, enhanced amyloid and tau aggregation, and breakdown of the blood–brain barrier. The outer ring aligns specific therapeutic strategies with each dysfunction, illustrating how APOE4’s diverse molecular effects require equally diverse intervention points. The figure thus serves as a conceptual roadmap for developing APOE4-targeted therapies grounded in mechanistic insight.
Taken together, the article argues for a shift in how Alzheimer’s disease—especially in APOE4 carriers—is understood and treated. Rather than viewing APOE4 solely as a genetic risk marker, the field is increasingly recognizing it as a central biological driver that shapes lipid metabolism, inflammation, vascular health, and neuronal resilience. By addressing these upstream mechanisms in parallel with more traditional amyloid- and tau-directed approaches, it may become possible to intervene earlier, reduce disease progression, and ultimately offer more effective and personalized care for individuals at heightened genetic risk.
This article appears as part of the Symposium Proceedings on APOE-Targeted Therapeutics, co-chaired by Hussein Yassine and Ornit Chiba-Falek, who helped lead and integrate discussions across multiple laboratories and research perspectives. The work represents a collaborative effort contributed by Cristelle Hugo, Bernadette O’Donovan, Isaiah O. Stephens, Lance A. Johnson, Gregory Cole, Julia TCW, Jan Johansson, and Kassandra Kisler, whose research programs span lipid biology, neuroinflammation, vascular physiology, molecular therapeutics, and human genetics.
.